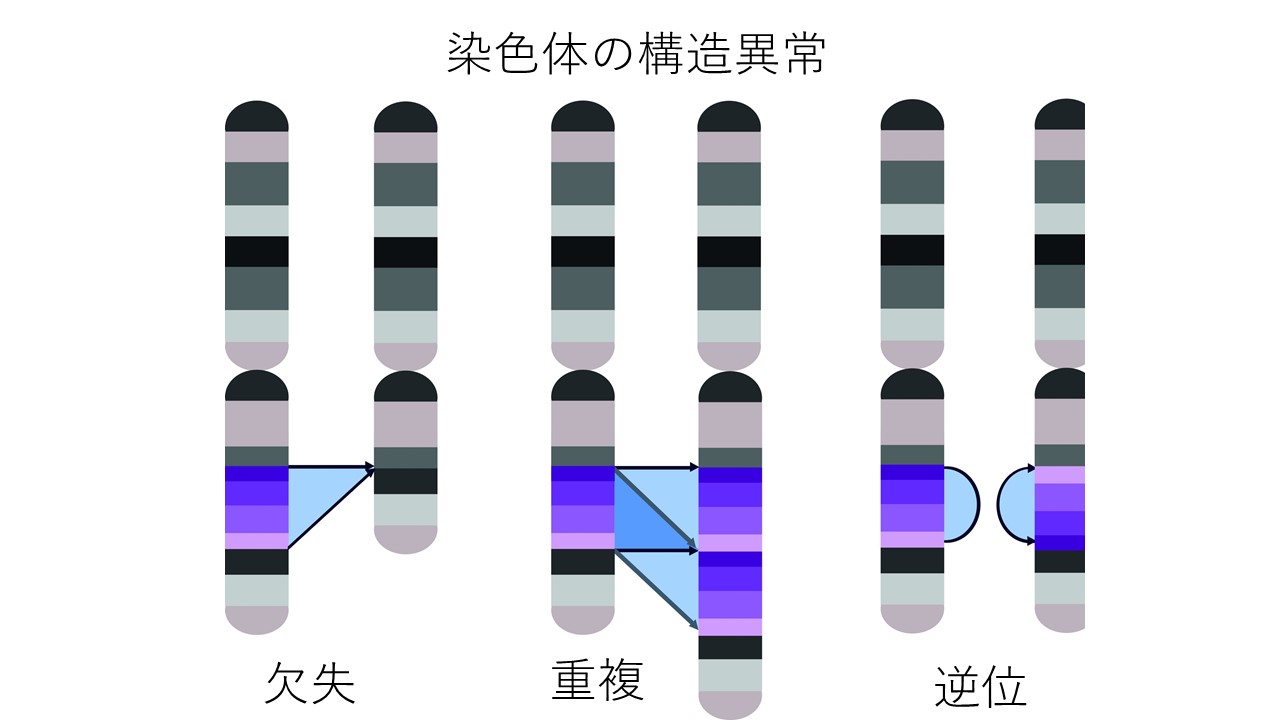
微小欠失症候群(微細欠失症候群)とは?
微小欠失症候群とは、染色体の特定の領域に存在する遺伝子群が欠失することによって引き起こされる一群の遺伝性疾患です。この欠失は超顕微鏡的なサイズであり、通常は50万塩基から300万塩基の範囲内で発生します. 微小欠失症候群は、重度の先天奇形や知的障害、身体的成長の障害などを引き起こす可能性があります.
微小欠失症候群には様々なタイプがあり、それぞれ異なる染色体の領域が関与しています。例えば、22q11.2欠失症候群は22番染色体のq11.2領域の欠失によって生じ、先天性心疾患や特徴的な顔貌、免疫系の問題などを引き起こします. 1p36欠失症候群は1番染色体の短腕末端の1p36領域の欠失により、成長障害や精神発達遅滞、難治性てんかんなどの症状が現れます.
これらの症候群は、通常の染色体検査では見つけにくいため、蛍光in situハイブリダイゼーション(FISH)やマイクロアレイ解析などのより高度な遺伝学的検査によって診断されることが多いです. 微小欠失症候群は母体の年齢に関係なく自然発生するものと考えられており、特定の遺伝子の欠失が原因であることが多いですが、家族内での遺伝の可能性もあります.
治療に関しては、染色体の異常そのものを治療する方法はありませんが、合併症に対する治療方法は存在します。例えば、心疾患に対しては、新生児期から体質に合わせた心臓手術計画を立て、一生にわたって臨床症状に基づいた治療や生活指導を受ける必要があります.
染色体微小欠失とは
少なくとも5メガベース(Mb)以上の染色体欠失は、通常、染色体バンド型の核型上で顕微鏡で確認することができます。こうした通常の細胞遺伝学で見えるはずの大きな欠失や重複は、サブ染色体異常(サブクロモゾーマルな異常)と呼ばれることもあります。
サブクロモゾーマルに対して、微小欠失、または亜微小欠失とは、従来の細胞遺伝学的手法を用いて光学顕微鏡で検出するには小さすぎる染色体欠失のことです。5Mbあれば光学顕微鏡で見えるわけですから、微小欠失とは5Mb未満の欠失を呼びます。これらの欠失を同定するためには、そのサイズに応じた専門的な検査が必要となります。微小欠失は通常1~3Mbの長さで、複数の連続した遺伝子が関与しています。症候群を引き起こす微小欠失の正確な大きさや位置は様々ですが、特定の「臨界領域」が一貫して関与しています。これらの微小欠失のほとんどの表現型への影響は、いくつかの重要な遺伝子のハプロ不全(2対ある遺伝子の片方が機能不全となること)、または場合によってはその病原性は単一の遺伝子のハプロ不全により惹起されます。
これらは通常、de novo(新生突然変異)であり、低コピー繰り返し配列(LCR)の隣接する相同組換えにより、同じ領域で再発する傾向があります。
欠失とは何ですか?何がなくなるのですか?
遺伝医学では欠失とは、DNAの複製中に染色体の一部またはDNAの配列がコピーされずになくなってしまう突然変異をいいます。欠失するサイズは1塩基から染色体全体に至るまでとさまざまです。微細欠失はこのうち5Mb 未満の欠失と定義されています。
微細欠失はどのくらいの頻度で起こるのでしょうか?
トリソミー、モノソミーといった染色体異数性(染色体の数が染色体単位で増える減る)とは異なり、最も一般的な微細欠失症候群や微細重複症候群は母体の年齢とは関係がありません。
染色体微小欠失の発生頻度は、特定の症候群によって異なります。例えば、22q11.2欠失症候群は約4,000~5,000人に1人の頻度で発生するとされています。一方で、Y染色体の微小欠失は、無精子症または重度乏精子症の男性において約5-10%の頻度で認められると報告されています. また、ウォルフヒルシュホーン症候群は約1/50,000人程度と推定されていますが、実際にはもっと多い可能性があるとも考えられています。
臨床的に関連のある微小欠失と重複は構造的に正常な妊娠の1.7%に発生します。
微細欠失と染色体異数性(トリソミー)の一番の違いは何でしょうか?
微小欠失は、染色体の一部が欠損してしまうだけなので、トリソミーでは大部分が流産死産になるのですが、微小欠失ではこれがあることだけによる流産死産はないと考えられることが一番大きな違いです。また、トリソミーに代表される染色体異数性は女性の高齢が危険因子(なりやすい)ことがわかっていますが、微小欠失症候群(微細欠失症候群)は男性女性どちらからもおこりますし、年齢も人種や民族性も全く関係ありません。つまり、微小欠失症候群には危険因子がありません。
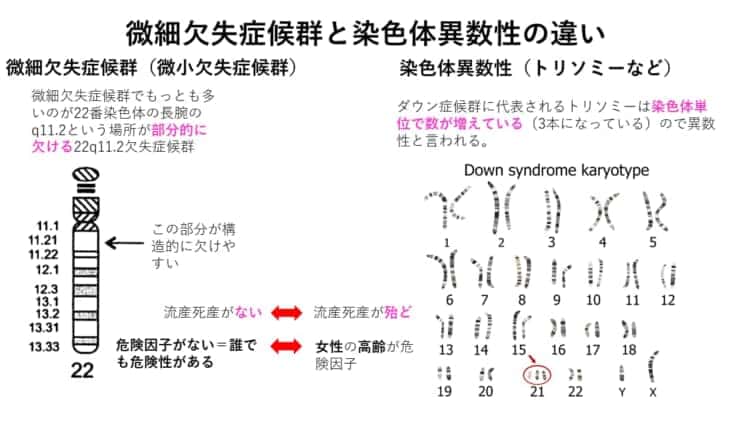
微小欠失症候群のお子さんを妊娠しやすい条件(危険因子)はあるのでしょうか?
たとえばダウン症候群のようなトリソミーでは女性が35歳以上であることが危険因子となることは有名です。
しかし、微小欠失症候群や微小重複症候群では危険因子は全くありません。ランダムに発生するのが特徴です。人種、性別、年齢全く関係ない積算リスクが1.7%というのは意外と高く感じませんか?
ただし。父母の一方が、何の症状もないけれども微小欠失を持っている場合には、1/2の確率でお子さんに遺伝します。その場合、親御さんには症状がなくても、お子さんに重篤な障害が発生することがあります。
微小欠失のあるお子さんが生まれた場合、親御さんがもっていないかどうかを検査することは重要になります。遺伝カウンセリングの現場では、一人目なら親御さんの検査をしない、二人目以降続く場合には検査する、という風習があるのですが。二人続いてから検査されて親にあったよねということになるよりは、一人目の時に微小欠失があると分かったのであれば、親御さんの検査をしておくべきではないかと、最近の私は考えてしまいます。
ある珍しい微小欠失のお子さんが二人続き、親御さんを調べて片方にあったと分かったご家族のお話を聞いて、考えを改めました。今は検査できるのだから、二人目がそうなら確率は高まるよね、ではなく、検査して次のお子さんのリスクをはっきりさせるべきじゃないかと思います。
微小欠失症候群は遺伝するのでしょうか?
微小欠失症候群のお子さんが親御さんになると常染色体優性遺伝形式で遺伝します。
現在までに報告されている微小欠失症候群はいくつくらいあるのですか?
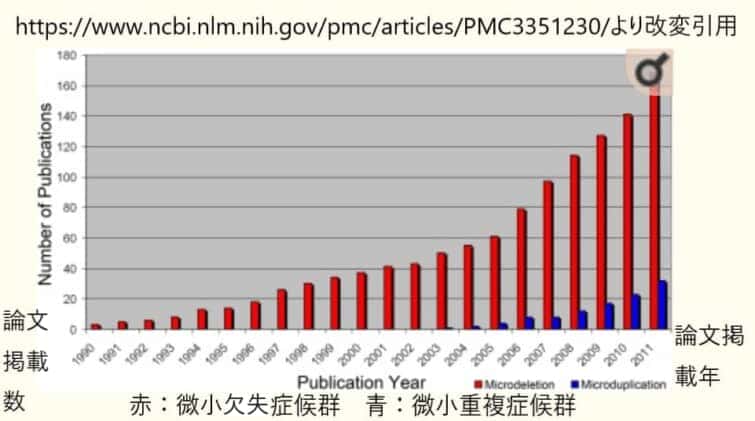
昔は診断すること自体が難しかったのですが、マイクロアレイなどの検出方法が開発されて増えてきました。
300種弱の微細欠失症候群・微細重複症候群が現在までに報告されています。
ゲノム疾患の概要
ゲノム障害とは、染色体/DNA物質の喪失または獲得に起因する疾患である。最も一般的で、より明確に定義されているゲノム障害は、コピー数の喪失(欠失症候群)とコピー数の増加(重複症候群)に起因するものとの2つの主要なカテゴリーに分けられます。詳細はゲノム疾患のページをご覧ください。
コピー数変動(CNV)とは
コピー数変動(CNV)とは、DNAの1つ以上の部分のコピー数におけるゲノムの微視的な違いで、DNAの獲得または喪失をもたらすものです。
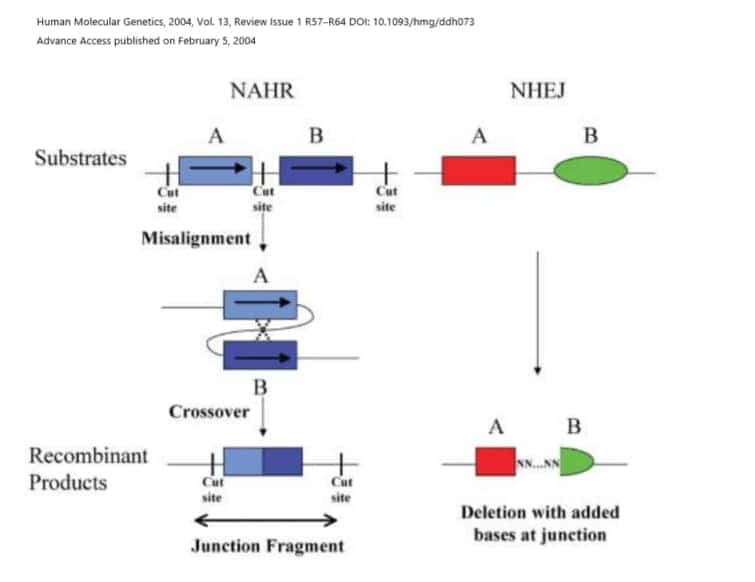
いくつかのCNVは病原性があり、ここで議論されているように、一貫した表現型の特徴を持つ症候群性の障害を引き起こします。他のCNVは疾患感受性または抵抗性と関連しており、同じCNVはいくつかの多様な疾患と関連している可能性があります。また、CNVには正常な遺伝的変異の一部であり、疾患との関連性が認められていないものもあります。隣接遺伝子症候群は、CNVが複数の隣接遺伝子に影響を及ぼす場合に起こりうるものです。
ゲノム障害における疾患を引き起こす主なメカニズムは、欠失または重複による線量感受性遺伝子または遺伝子のコピー数の変化です。このため、遺伝子が機能しなくなり、タンパクの産生量が減ります。他の疾患メカニズムとしては、良い方のアレルが不活化されることでたまたま対立遺伝菓子が劣性の病的変異だったため劣性疾患が発症してしまう、インプリンティング遺伝子では父母どちらかの遺伝子が組織特異的に発現するのですがそれが病的になり発症する、遺伝子ではない部分にある調節エレメントがおかしくなる、などがあります。
ゲノム障害の検出方法
ゲノム障害は通常、アレイ比較ゲノムハイブリダイゼーション(CGH)によって検出されます。ほとんどの検査室では、蛍光in situハイブリダイゼーション(FISH)、多重ライゲーション依存性プローブ増幅(MLPA)、定量的ポリメラーゼ連鎖反応(Q-PCR)などの独立した方法で、アレイ上で検出されたゲインまたはロスを確認しています。
現在までに報告されている微小欠失症候群
詳細は各リンク先をご覧ください。ここでは欠失サイズの大きさのみ言及します。(疾患詳細は順番にページをおつくりいたします)
1p36欠失症候群: 2.2 ~ 10.2 Mb
1q21.1遠位微小欠失症候群: 1.35 Mb
1q21.1近位微小欠失症候群: 200kb (0.2Mb)
1q43-44微小欠失症候群 : ~3.5Mb
2p15-16.1微小欠失症候群: 4~6Mb
2q23.1微小欠失症候群 : 2.4~5.4 Mb
2q33.1微小欠失症候群(GLASS症候群) : 35 kb(0.035Mb)~10.4 Mb
2q37微小欠失症候群 : 3.5~8.8Mb
3pter-p25微小欠失症候群 : 10.2~11.0Mb
3q29微小欠失症候群 : 1.6Mb
4p微小欠失症候群 (WOLF-HIRSCHHORN症候群) : 200Kb(0.2Mb)~34.0Mb
5q35微小欠失症候群 (SOTOS症候群) : ~2.0 Mb
6p25微小欠失症候群 : 0.9~11.5Mb
7q11.23微小欠失症候群(WILLIAMS症候群) : 1.5-1.8Mb
8q22.1微小欠失症候群 (仮面様顔症候群) : 1.6~7.2Mb
8q24.11微小欠失症候群(LANGER-GIEDION症候群):95kb(0.09Mb)~9Mb
9p22微小欠失症候群 : 5~8Mb
9q34.3微小欠失症候群(9qサブテロメア欠失症候群): 700 kb(0.7Mb) ~ 2.3 Mb
10p14-p13微小欠失症候群(DiGEORGE症候群2型):2Mb
11p13微小欠失症候群(WAGR症候群): 0.6Mb
11p11.2微小欠失症候群(POTOCKI-SHAFFER症候群): ~1.0Mb
11q24.1微小欠失症候群(JACOBSEN症候群): 1.5~12.8Mb
13q14微小欠失症候群(網膜芽細胞腫症候群): 4~10Mb
15q11.2微小欠失症候群
15q11-13微小欠失症候群 母系:ANGELMAN症候群 父系:PRADER-WILLI症候群(PWS):5~7Mb
15q13.3微小欠失症候群:1.5Mb
15q15.3微小欠失症候群
15q24微小欠失症候群 : 1.7 ~ 3.9 Mb
16p13.3微小欠失症候群(RUBINSTEIN-TAYBI症候群):
16p13.11微小欠失症候群:1.65Mb
16p12.2微小欠失症候群
16p11.2微小欠失症候群 : 29.5~30.1Mb
17p13.3微小欠失症候群
17p11.2微小欠失症候群 (遺伝性圧脆弱性ニューロパチー)
17p11.2微小欠失症候群 (SMITH-MAGENIS症候群)
17q12微小欠失症候群 :1.5Mb
17q21.31微小欠失症候群 :
18p微小欠失症候群 :
20p11微小欠失症候群 (ALAGILLE症候群):
22q11.2微小欠失症候群 :3Mb
遠位22q11.2微小欠失症候群 :1.4~2.1Mb
22q13.3微小欠失症候群(PHELAN-MCDERMID症候群):

この記事の監修・執筆者:仲田 洋美
(臨床遺伝専門医/がん薬物療法専門医/総合内科専門医)
ミネルバクリニック院長。1995年に医師免許を取得後、
臨床遺伝学・内科学・腫瘍学を軸に診療を続けてきました。
のべ10万人以上のご家族の意思決定と向き合ってきた臨床遺伝専門医です。
出生前診断(NIPT・確定検査・遺伝カウンセリング)においては、
検査結果の数値そのものだけでなく、
「結果をどう受け止め、どう生きるか」までを医療の責任と捉え、
一貫した遺伝カウンセリングと医学的支援を行っています。
ハイティーンの時期にベルギーで過ごし、
日本人として異文化の中で生活した経験があります。
価値観や宗教観、医療への向き合い方が国や文化によって異なることを体感しました。
この経験は現在の診療においても、
「医学的に正しいこと」と「その人にとって受け止められること」の両立を考える姿勢の基盤となっています。
また、初めての妊娠・出産で一卵性双生児を妊娠し、
36週6日で一人を死産した経験があります。
その出来事は、妊娠・出産が女性の心身に与える影響の大きさ、
そして「トラウマ」となり得る体験の重みを深く考える契機となりました。
現在は、女性を妊娠・出産のトラウマから守る医療を使命の一つとし、
出生前診断や遺伝カウンセリングに取り組んでいます。
出生前診断は単なる検査ではなく、
家族の未来に関わる重要な意思決定です。
年齢や統計だけで判断するのではなく、
医学的根拠と心理的支援の両面から、
ご家族が後悔の少ない選択をできるよう伴走することを大切にしています。
日本人類遺伝学会認定 臨床遺伝専門医/日本内科学会認定 総合内科専門医/
日本臨床腫瘍学会認定 がん薬物療法専門医。
2025年には APAC地域における出生前検査分野のリーダーとして国際的評価を受け、
複数の海外メディア・専門誌で特集掲載されました。

