目次
Q. DiGeorge症候群(22q11.2欠失症候群)とはどのような病気ですか?
A. 22番染色体の22q11.2領域が欠失することで、心臓(円錐動脈幹)・免疫(胸腺)・副甲状腺(低カルシウム血症)などに影響が出る先天性疾患です。
症状の幅が広く、軽症〜重症(先天性無胸腺)までスペクトラムがあります。
- ➤代表的三徴:心疾患(円錐動脈幹)・胸腺低形成・低カルシウム血症
- ➤原因:22q11.2の微小欠失(多くは新生突然変異)
- ➤遺伝形式:常染色体優性(顕性)(ただし表現型は多様)
- ➤確定診断:染色体マイクロアレイ(CMA)が標準
- ➤重症免疫不全:先天性無胸腺(complete DGS)は早期介入が重要
1. DiGeorge症候群(22q11.2欠失)とは|基本情報
【結論】 DiGeorge症候群は、胚発生の咽頭弓・咽頭嚢(pharyngeal pouch)系の発達障害により、心臓・胸腺・副甲状腺などに異常が起こる疾患群です。多くは22q11.2欠失が原因で、一般に22q11.2欠失症候群(22q11.2DS)と呼ばれます。
症状は非常に幅広く、新生児期に重篤な心疾患や低カルシウム血症で見つかる場合もあれば、学童期以降に学習・発達・反復感染を契機に診断されることもあります。
💡 用語解説:「先天性無胸腺(congenital athymia)」とは?
胸腺がほとんど存在しない(または機能しない)状態で、T細胞が作れず、重症複合免疫不全(SCID)に近い免疫不全となります。22q11.2DSの中では少数(約0.5〜1%)ですが、早期の専門的対応が必要です。
💡 用語解説:「咽頭弓(いんとうきゅう)・咽頭嚢(いんとうのう)」とは?
胎児の発生初期に、のど(咽頭)周囲に一時的に作られる「顔・首・心臓の材料」となる構造です。咽頭弓は外側のふくらみ(アーチ)で、ここから顎・耳・顔の一部、心臓の大血管(大動脈弓)などが形づくられます。咽頭嚢は内側の袋状の構造で、ここから胸腺(免疫)や副甲状腺(カルシウム調節)が発達します。DiGeorge症候群ではこの発達過程がうまく進まず、心疾患・免疫異常・低カルシウム血症といった症状につながります。
疾患の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | 22q11.2欠失症候群(DiGeorge症候群/VCFSを含む概念) |
| 主な特徴 | 円錐動脈幹心疾患、胸腺低形成、低カルシウム血症、口蓋異常、発達・精神症状など |
| 遺伝形式 | 常染色体優性(顕性)(多くは新生突然変異) |
| 重症免疫不全 | 先天性無胸腺(complete DGS)は稀だが生命予後に直結 |
2. 主な症状|心臓・免疫・低カルシウム血症
【結論】 22q11.2欠失症候群は、心疾患、免疫異常、低カルシウム血症を中心に、口蓋・耳鼻科・腎尿路・発達/精神など多臓器に影響します。症状の組み合わせは人により大きく異なります。
心疾患(円錐動脈幹)
| 代表的心疾患 | ポイント |
|---|---|
| 大動脈弓離断 | 新生児期に循環不全で発見されやすい |
| 総動脈幹症 | チアノーゼ性心疾患、早期治療が重要 |
| ファロー四徴症 | 22q11.2DSとの関連がよく知られる |
| VSD/ASD、血管輪など | 軽症〜呼吸・嚥下症状まで幅広い |
低カルシウム血症(副甲状腺低形成)
新生児期にけいれん・テタニーで発見されることがあります。血液検査では低カルシウム、高リン、PTH低値が典型です。成人になってからも、強いストレス(手術・出産・感染など)で低カルシウムが再燃することがあります。
免疫異常(胸腺低形成〜先天性無胸腺)
- ➤多くは軽〜中等度のT細胞減少で、重篤な免疫不全ではない
- ➤一部で反復する呼吸器感染、自己免疫、アレルギーが問題になる
- ➤ごく一部に先天性無胸腺(complete DGS)があり、早期の免疫再構築が必要
3. 原因と遺伝的背景|TBX1と欠失の起こり方
【結論】 22q11.2欠失の臨床像の中心にはTBX1(転写因子)を含む遺伝子群のハプロ不全が関与します。欠失は、22q11.2領域にある反復配列(SD/LCR)同士の誤った組み換えで起こりやすいことが知られています。
欠失が起きやすい理由(NAHR)
💡 用語解説:「非対立遺伝子間相同組換え(NAHR)」とは?
本来、減数分裂では同じ位置(アレル)同士で組み換えが起こりますが、22q11.2のようによく似た配列(SD/LCR)が複数あると、別の位置の配列同士で誤って組み換えが起こることがあります。これにより欠失や重複が生じます。
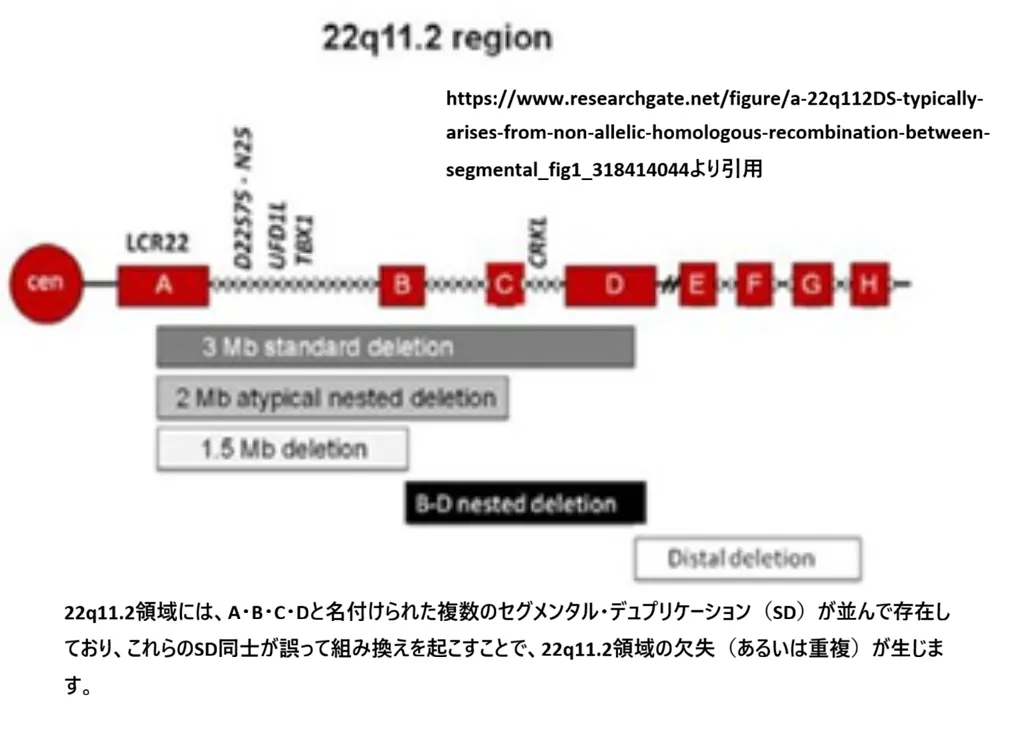
※図:22q11.2領域のSD(A〜D)と組み換えによる欠失の概念(著者改変)

🩺 院長コラム【“欠失がある=同じ経過”ではありません】
22q11.2欠失症候群は、同じ欠失サイズでも症状の出方が大きく異なります。心臓が重いが免疫は軽い方もいれば、その逆もあります。
出生前診断や小児期の検査で見つかった場合、私が必ず強調するのは「今わかっている事実」と「まだ予測できない部分」を分けて考えることです。遺伝カウンセリングでは、検査の限界や不確実性も含めて、家族の意思決定を支援します。
4. 診断方法|新生児から成人まで
【結論】 22q11.2欠失の確定診断は染色体マイクロアレイ(CMA)が標準です。FISHやMLPAは特定領域の確認に有用ですが、CMAは網羅的にCNVを評価できます。
新生児で疑う場面
- ➤円錐動脈幹心疾患(ファロー四徴症、総動脈幹症、大動脈弓離断など)
- ➤原因不明の低カルシウム血症(けいれん/テタニー)
- ➤口蓋裂・鼻咽腔閉鎖不全、反復感染、T細胞減少
出生前・確定検査での位置づけ
| 検査 | 位置づけ | ポイント |
|---|---|---|
| NIPT | △ スクリーニング | 結果解釈には検査性能・偽陽性/偽陰性を踏まえた説明が必要 |
| 羊水検査+CMA | ◎ 確定診断 | Gバンド法では検出できない微小欠失を確定診断可能。 ※学会指針では、原則として超音波での構造異常がある場合などが対象とされています。 |
| 絨毛検査+CMA | ◎ 確定診断 | 妊娠初期に評価可能(施設・適応による) |
似た疾患との比較|疑うポイントと検査の道筋
22q11.2欠失症候群は、他の微小欠失/重複とも合併や症状が重なる部分があります。下表で“症状の重なり→推奨される検査”を整理します。
| 疾患/領域 | 疑うサイン | よく見られる合併 | 検査の道筋 | 関連リンク |
|---|---|---|---|---|
| 22q11.2欠失症候群 | 円錐動脈幹心疾患、低カルシウム血症、免疫異常(胸腺低形成) | 口蓋裂/鼻咽腔閉鎖不全、反復感染、発達/学習/精神症状 | CMA(染色体マイクロアレイ):確定 | 22q11.2欠失 |
| 1p36欠失症候群 | 重度発達遅滞、筋緊張低下、てんかん | 心疾患、感覚/摂食障害など | CMA:確定 | 1p36欠失 |
| 15q11.2 CNV | 発達/行動、学習の遅れ | 行動特性、軽度の認知差など | CMA:確定 | 15q11.2 CNV |
| 15q12(PWS) | 乳児期の筋緊張低下、過食/肥満 | 低身長、行動特性 | メチル化検査が中核+CMA | 15q12(PWS) |
| 15q12(Angelman) | 重度言語障害、てんかん | 運動失調、睡眠/行動特性 | メチル化検査が中核+CMA | 15q12(AS) |
| 16p11.2 欠失/重複 | ASD/発達・言語、体格差(欠失/重複) | 頭囲偏差、行動特性、てんかん | CMA:確定 | 16p11.2 領域 |
⚠️ 解釈上の注意:似ている症状があっても、症状だけで特定するのは誤解の元です。まずはCMA(染色体マイクロアレイ)で網羅的に評価し、必要に応じて
メチル化検査(PWS/ASのようなインプリンティング系)などを追加してください。
5. 治療と長期管理|多職種連携が基本
【結論】 22q11.2欠失症候群は多臓器にまたがるため、循環器・内分泌・免疫・耳鼻科/口蓋・発達/精神などの多職種連携が重要です。重症免疫不全(先天性無胸腺)が疑われる場合は、早期に専門施設での免疫再構築(胸腺移植など)を検討します。
- ➤心疾患:術後も含めた循環器フォロー
- ➤低カルシウム:乳児期だけでなくストレス時再燃に注意
- ➤免疫:T細胞数・ワクチン反応・反復感染の評価
- ➤発達/精神:学童期以降の学習支援、思春期以降の精神症状の早期把握
🩺 院長コラム【“治療”より先に“整理”が大切です】
22q11.2欠失症候群の診療では、いきなり「何をすべきか」を決める前に、今ある問題(心臓・免疫・カルシウム・発達など)を整理し、優先順位をつけることが重要です。
同じ診断でも必要なフォローは人によって全く異なります。私は、家族が「やるべきことが多すぎて混乱する」状態にならないよう、“短期の安全”と“長期の見通し”を分けて説明することを大切にしています。
6. 遺伝カウンセリング|家族への説明と再発リスク
【結論】 22q11.2欠失症候群は表現型が非常に多様で、将来予測にも幅があります。遺伝カウンセリングでは、疾患の特徴に加え、家族への検査(両親の欠失の有無)と再発リスクの整理が重要です。
当院では遺伝カウンセリングとはの考え方に基づき、臨床遺伝専門医が説明を担当します。
| 状況 | 次子への再発リスク |
|---|---|
| 両親とも欠失なし(新生突然変異が示唆) | 一般に低い(ただし生殖細胞モザイクの可能性はゼロではない) |
| 片親が欠失を保有 | 50%(常染色体優性[顕性]) |
7. 出生前診断|NIPTと羊水検査・絨毛検査
【結論】 22q11.2欠失は出生前検査で指摘されることがありますが、NIPTはスクリーニングであり、確定には羊水検査または絨毛検査でのCMAが必要です。結果の受け止め方はご家族ごとに異なるため、丁寧な遺伝カウンセリングが欠かせません。
ミネルバのNIPT(ダイヤモンドコース)で評価できる微小欠失
当院のダイヤモンドコースでは、微小欠失を12箇所評価します(22q11.2欠失を含む)。
- ➤1p36 del / 2q33 del / 4p16 del / 5p15 del
- ➤8q23q24 del / 9p del / 11q23q25 del / 15q11.2-q13 del
- ➤17p11.2 del / 18p del / 18q22q23 del / 22q11.2 del
確定検査と費用面の安心
確定診断が必要な場合は、羊水検査・絨毛検査の料金説明もご参照ください。当院のNIPT受検者には、互助会制度(互助会費は8,000円。NIPT受検者全員に適用)があるため、陽性時の羊水検査費用を全額補助します。
一人で悩まず、専門医を頼ってください
22q11.2欠失(DiGeorge症候群)について不安がある方は、
臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
よくある質問(FAQ)
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
参考文献
- [1] McDonald-McGinn DM, et al. 22q11.2 deletion syndrome. [PubMed]
- [2] Scambler PJ. The 22q11 deletion syndromes. [PubMed]
- [3] Bassett AS, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. [PubMed]
- [4] Markert ML, et al. Thymus transplantation for congenital athymia (complete DiGeorge). [PubMed]
- [5] GeneReviews: 22q11.2 Deletion Syndrome. [NCBI Bookshelf]
- [6] OMIM: 22q11.2 deletion syndrome. [OMIM]
- [7] DECIPHER: 22q11.2 recurrent deletion. [DECIPHER]