目次

📍 クイックナビゲーション
Q. NIPTで8トリソミーが陽性でした。赤ちゃんは大丈夫ですか?
A. 第1〜8番染色体の完全トリソミーはほとんどが初期流産となり、出生に至ることは極めて稀です。NIPTで陽性の場合、多くは限局性胎盤モザイク(CPM)やバニシングツインが原因で、胎児自体は正常なケースが多いです。ただし、確定検査で正確な状態を把握することが重要です。
-
➤
第1〜8番染色体の完全トリソミー → ほぼ100%が初期流産(出生例は極めて稀) -
➤
8トリソミーの特異性 → モザイク型のみ生存可能(ワーカニー症候群2型) -
➤
NIPT陽性の主な原因 → 限局性胎盤モザイク(CPM)、バニシングツイン、母体腫瘍など -
➤
関連する微細欠失症候群 → 1p36欠失、ウォルフ・ヒルシュホーン、猫鳴き、ウィリアムズ症候群など -
➤
ミネルバの強み → 確定検査まで院内完結、臨床遺伝専門医が一貫対応
1. 第1〜8番染色体とは?大型染色体の特徴
【結論】 第1〜8番染色体は、ヒトの染色体の中でも最も大きく、遺伝子数も多い「大型染色体」です。そのため、これらの染色体に数的異常(トリソミーやモノソミー)が生じると、発生に致命的な影響を与えます。
「NIPTで聞き慣れない染色体の異常が指摘された」「8トリソミーって何?」そんな不安を抱えていらっしゃるかもしれませんね。まずは、第1〜8番染色体がどのような特徴を持つのか、一緒に確認していきましょう。
💡 用語解説:トリソミーとは?
通常、人間の細胞には各染色体が2本ずつ(父親由来1本、母親由来1本)存在します。トリソミーとは、特定の染色体が3本になった状態のことです。21番染色体が3本になる「21トリソミー」がダウン症候群として知られています。
染色体のサイズと遺伝子数
ヒトの染色体は1番から22番まで、基本的にサイズの大きい順に番号がつけられています。第1番染色体はヒトゲノムの約8%を占める最大の染色体で、約3,000個以上の遺伝子を含んでいます。
| 染色体 | サイズ(Mb) | 推定遺伝子数 | 完全トリソミーの予後 |
|---|---|---|---|
| 1番 | 約249 | 約3,000+ | 致死的(出生例なし) |
| 2番 | 約243 | 約2,500+ | 致死的(流産の1〜2%) |
| 3番 | 約198 | 約1,100+ | 致死的 |
| 4番 | 約191 | 約1,000+ | 致死的(流産の2〜3%) |
| 5番 | 約181 | 約900+ | 致死的 |
| 6番 | 約171 | 約1,000+ | 致死的(UPDリスクあり) |
| 7番 | 約159 | 約1,000+ | 致死的(CPM多い、UPDリスク) |
| 8番 | 約146 | 約700+ | モザイク型のみ生存可能 |
比較として、ダウン症候群の原因である21番染色体は、最も小さな染色体の一つで、約200〜300個の遺伝子しか含まれていません。染色体が小さく遺伝子数が少ないからこそ、トリソミーでも出生が可能なのです。
2. なぜ第1〜8番染色体の完全トリソミーは致死的なのか
【結論】 第1〜8番染色体は遺伝子数が多いため、トリソミーになると遺伝子量効果(Gene Dosage Effect)により、細胞の正常な機能が破綻します。その結果、着床前後〜妊娠初期に淘汰されます。
「なぜダウン症は生まれてくるのに、8トリソミーは生まれないの?」という疑問を持つ方もいらっしゃるでしょう。その理由は、染色体のサイズと遺伝子量にあります。
💡 用語解説:遺伝子量効果とは?
トリソミーでは、その染色体上のすべての遺伝子が1.5倍量発現します。遺伝子が多い大型染色体では、この「過剰発現」が細胞周期の制御、代謝、発生シグナルを大きく狂わせ、胚発生が維持できなくなります。これを遺伝子量効果と呼びます。
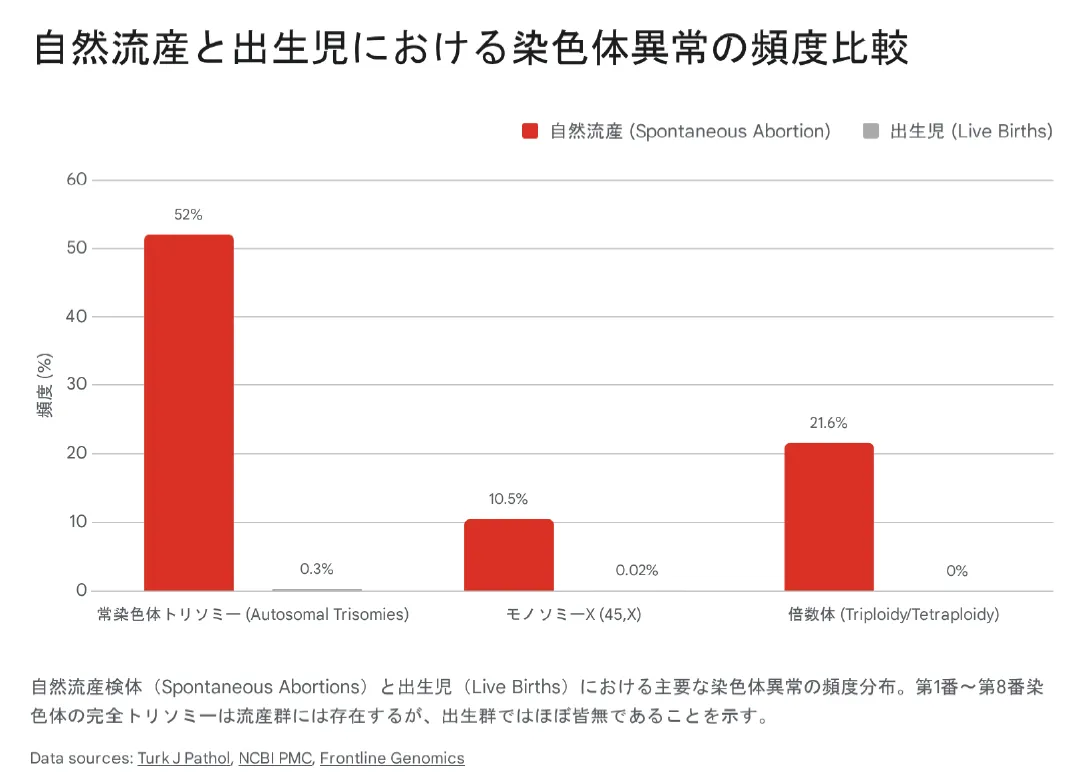
流産における染色体異常の頻度
自然流産の約50〜60%には染色体異常が関係しています。中でも、常染色体トリソミーは最も多い原因です。
-
•
自然流産における常染色体トリソミー:約52%
-
•
出生児における常染色体トリソミー:約0.3%(主に13, 18, 21番)
-
•
第1〜8番染色体の完全トリソミー:出生例はほぼ0%
-
•
重要な事実:染色体が大きいほど、トリソミーは致死的

🩺 院長コラム【「致死的」という言葉に怖がらないでほしい】
「致死的」という言葉を聞くと、とても怖く感じるかもしれません。でも、これは自然の「生物学的フィルター」が正常に機能している証拠でもあるのです。
第1〜8番染色体の完全トリソミーは、ほとんどが妊娠に気づく前の着床前後で淘汰されます。お母さんの体が「知らないうちに」守っているのです。
NIPTで陽性が出た場合も、胎児自体が本当にその異常を持っているとは限りません。胎盤だけに異常がある「限局性胎盤モザイク」や、消えた双子の「バニシングツイン」が原因であることも多いのです。だからこそ、確定検査で正確な情報を得ることが大切です。
3. 8トリソミー(ワーカニー症候群2型)の特異性
【結論】 第1〜8番染色体の中で、8トリソミーは唯一、モザイク型で出生・生存が可能な特異的な染色体異常です。これは「ワーカニー症候群2型(Warkany Syndrome 2)」として確立された症候群です。
8トリソミーは、第1〜8番染色体の異数性の中で最も研究が進んでいる異常です。なぜなら、モザイク型であれば出生後も長期生存が可能であり、臨床的に明確な症候群として定義されているからです。
💡 用語解説:モザイク型とは?
モザイク型とは、体の一部の細胞だけが異常な染色体を持ち、残りの細胞は正常な状態です。正常な細胞が混在することで、完全トリソミーよりも遺伝子量効果が軽減され、生存が可能になります。モザイク率(異常細胞の割合)によって症状の重さが異なります。
ワーカニー症候群2型の特徴
-
•
最も特徴的な所見:手のひらと足底の非常に深い掌紋・足底紋(「ひだ」のように深く刻まれた皺)
-
•
発生頻度:約1/25,000〜1/50,000出生
-
•
男女比:約5:1で男性に多い
-
•
顔貌:長く細い顔、広い鼻根、厚く外翻した下唇、大きな異形成耳
-
•
骨格異常:関節拘縮、脊椎側弯、肋骨異常
-
•
知的発達:正常範囲から重度まで様々(モザイク率による)
-
•
重要なリスク:血液腫瘍(MDS、AML、CML)の発症リスク上昇
⚠️ 診断上の注意点:モザイク型8トリソミーには顕著な組織特異的淘汰があります。末梢血リンパ球では、加齢とともに正常細胞が優位になり、トリソミー細胞が消失する傾向があります。そのため、血液検査だけでは診断できないことがあり、皮膚線維芽細胞の検査が必要になる場合があります。
4. 各染色体に関連する主な症候群
【結論】 第1〜8番染色体の完全トリソミーは致死的ですが、部分的な欠失や重複(微細欠失症候群)では出生・生存が可能なものがあります。代表的なものに、1p36欠失症候群、ウォルフ・ヒルシュホーン症候群、猫鳴き症候群、ウィリアムズ症候群などがあります。
完全トリソミーとは別に、染色体の一部が欠けたり重複したりする「構造異常」があります。これらは微細欠失症候群として知られ、NIPTや羊水検査で検出できるものもあります。
| 染色体 | 主な症候群 | 頻度 | 主な特徴 |
|---|---|---|---|
| 1番 | 1p36欠失症候群 | 1/5,000〜1/10,000 | 重度知的障害、てんかん、先天性心疾患 |
| 2番 | 2q37欠失症候群 | 稀 | 短指症、軽〜中度知的障害、肥満傾向 |
| 3番 | 3p欠失症候群 | 稀 | 重度発達遅滞、小頭症、眼瞼下垂 |
| 4番 | ウォルフ・ヒルシュホーン症候群(4p-) | 1/50,000 | 「ギリシャ兵のヘルメット」様顔貌、難治性てんかん |
| 5番 | 猫鳴き症候群(5p-) | 1/37,000〜1/50,000 | 猫のような甲高い泣き声、小頭症、知的障害 |
| 6番 | 父性UPD6→一過性新生児糖尿病 | 稀 | 出生直後の高血糖、IUGR、巨舌症 |
| 7番 | ウィリアムズ症候群(7q11.23欠失) | 1/7,500〜1/10,000 | 「妖精様顔貌」、大動脈弁上狭窄、社交的性格 |
| 7番 | 母性UPD7→シルバー・ラッセル症候群 | 1/30,000〜1/100,000 | 重度の発育不全、逆三角形の顔貌、左右非対称 |
| 8番 | モザイク型8トリソミー(ワーカニー症候群2型) | 1/25,000〜1/50,000 | 深い掌紋・足底紋、関節拘縮、血液腫瘍リスク |
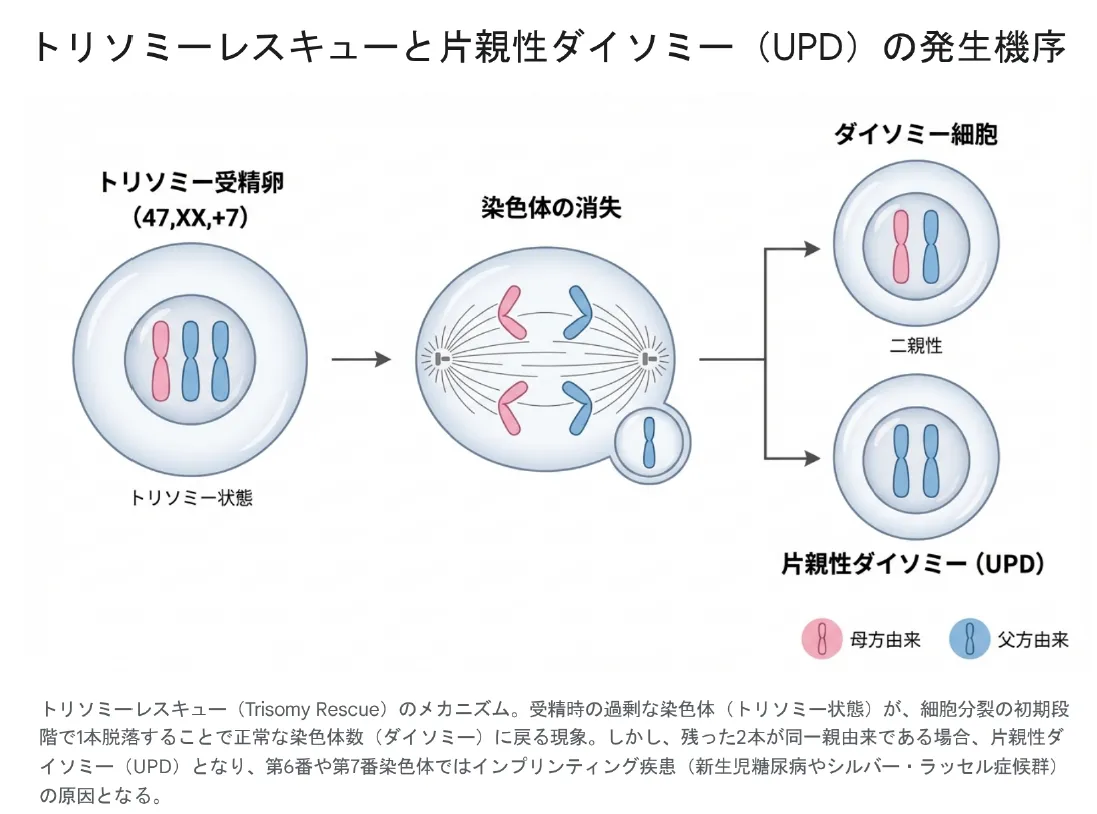
💡 用語解説:UPD(片親性ダイソミー)とは?
片親性ダイソミー(UPD)とは、ある染色体の2本が両方とも同じ親(父親または母親)由来である状態です。これは「トリソミーレスキュー」という現象で起こることがあります。トリソミーの受精卵から余分な染色体が1本脱落して正常な2本に戻る際、偶然2本とも同じ親由来になると、インプリンティング疾患(遺伝子の親由来特異的発現の異常)を引き起こすことがあります。
染色体異常について心配なことはありませんか?
ネットの情報だけで判断するのは難しいもの。
臨床遺伝専門医に直接相談することで、正確な情報と安心を得られます。
※オンライン診療も対応可能です
🧬 各染色体異常(トリソミー・部分モノソミー)の詳細
この記事で解説した第1〜8番染色体の異数性や微細欠失・重複による疾患、および予後については以下のリンクから詳細をご確認いただけます。
5. NIPTで第1〜8番染色体の異常はわかる?
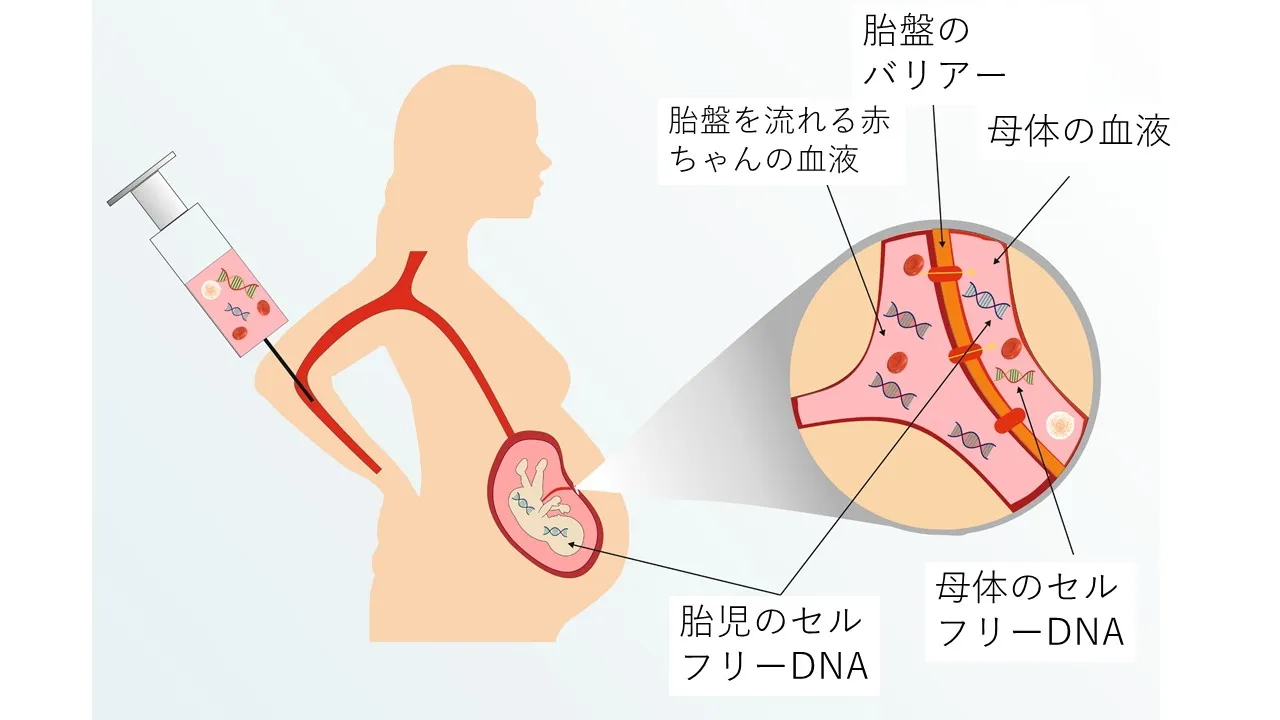
【結論】 NIPTの全染色体検査では、第1〜8番染色体のトリソミーも検出対象となります。ただし、陽性の場合は限局性胎盤モザイク(CPM)やバニシングツインの可能性が高く、必ず確定検査で胎児の状態を確認する必要があります。
「NIPTで8トリソミーが陽性と出たけど、赤ちゃんは本当に大丈夫?」そんな不安を抱えている方も多いでしょう。ここでは、NIPTで稀な染色体異常(Rare Autosomal Trisomies: RATs)が検出された場合の解釈について説明します。
NIPT陽性時に考えられる可能性
-
①
限局性胎盤モザイク(CPM)
胎盤組織のみにトリソミー細胞があり、胎児自体は正常なケース。NIPTは母体血中の胎盤由来DNAを検出するため、CPMでも陽性になります。最も多い原因です。 -
②
バニシングツイン(消失双胎)
妊娠初期に消えた双子の一方が異数性を持っていた場合、そのDNA断片が検出されます。現在の胎児は正常な場合があります。 -
③
母体の染色体異常
極めて稀ですが、母体の悪性腫瘍(がん細胞の染色体不安定性)由来のDNAが検出されることがあります。 -
④
胎児の真のトリソミー(モザイク)
第1〜8番の完全トリソミーは致死的ですが、モザイク型では稀に出生に至ることも(特に8トリソミー)。
⚠️ 重要:NIPTで第1〜8番染色体のトリソミーが陽性と出た場合でも、NIPT陽性のみで妊娠中断を決定することは絶対にしないでください。必ず羊水検査・絨毛検査による確定診断を行い、胎児の正確な状態を把握することが不可欠です。
CPMでも油断は禁物
胎児が正常であっても、CPMが存在すること自体がリスク因子となる場合があります。
CPMに伴うリスク
- •
胎盤機能不全による胎児発育不全(FGR)
- •
羊水過少
- •
早産リスクの上昇
- •
妊娠高血圧症候群
推奨される対応
- •
羊水検査で胎児の染色体を確認
- •
UPD検査(6, 7番の場合特に重要)
- •
胎児発育の慎重な経過観察
- •
ハイリスク妊娠として管理
6. 陽性時の対応と確定検査の重要性
【結論】 NIPTで稀な染色体異常(RATs)が陽性と出た場合、必ず確定検査(羊水検査・絨毛検査)で胎児の状態を確認することが必要です。特に6番・7番染色体の場合は、UPD(片親性ダイソミー)検査も検討されます。
「陽性と言われて頭が真っ白になった」「これからどうすればいいの?」そんな気持ちでいっぱいかもしれません。まずは深呼吸をして、一緒に次のステップを確認していきましょう。
陽性時の対応フロー
| ステップ | 内容 | 目的 |
|---|---|---|
| 1 | 遺伝カウンセリング | 結果の意味と次の選択肢を理解する |
| 2 | 羊水検査・絨毛検査 | 胎児の染色体を直接確認(確定診断) |
| 3 | UPD検査(必要に応じて) | 6, 7番染色体の場合、インプリンティング疾患のリスク評価 |
| 4 | 詳細超音波検査 | 胎児の構造異常の有無を確認 |
| 5 | 結果説明と今後の方針決定 | 正確な情報に基づいた意思決定 |
🩺 院長コラム【陽性結果を受け取ったあなたへ】
NIPTで稀な染色体異常の陽性結果を受け取ると、多くの方が「なんで私が」「赤ちゃんはどうなるの」と頭が真っ白になります。でも、陽性=赤ちゃんが異常、ではありません。
特に第1〜8番染色体のような「稀なトリソミー」の場合、胎盤だけに異常があって赤ちゃんは正常というケースが実は多いのです。だからこそ、焦って結論を出さないでほしいのです。
当院では、陽性結果が出た方には何度でも遺伝カウンセリングを受けていただけます。確定検査のご案内から結果の説明、そしてその後の選択肢まで、臨床遺伝専門医として最後まで寄り添います。一人で抱え込まないでください。
7. ミネルバクリニックの強み|確定検査まで院内完結
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。NIPTから確定検査(羊水検査・絨毛検査)まで院内で完結できる、稀有な体制を整えています。
🏥 確定検査まで院内完結
2025年6月より産婦人科を併設。羊水検査・絨毛検査も院内で実施可能。転院の必要なく、心理的負担を軽減できます。
👩⚕️ 臨床遺伝専門医が一貫対応
検査前のカウンセリングから結果説明、陽性時のフォローまで、臨床遺伝専門医が最初から最後まで担当します。
💰 互助会で費用面も安心
互助会(8,000円)に加入いただくと、陽性時の確定検査費用を全額カバー(上限なし)。安心して検査を受けられます。
💡 遺伝カウンセリング料金について
当院では遺伝カウンセリング料金33,000円が検査費用に内包されています。これは当日の説明だけでなく、陽性になった場合に何度でもカウンセリングを受けられること、妊娠経過中に心配なことがあればいつでも相談できることを含んでいます。「お金がかかるから相談しにくい」ということがないよう、なるべく安心して日常生活が送れるよう配慮しています。
よくある質問(FAQ)
🏥 一人で悩まないでください
NIPTの結果について心配なこと、検査を受けるかどうか迷っていること、
どんなことでもお気軽にご相談ください。
臨床遺伝専門医があなたとご家族に寄り添います。
📚 全染色体検査・完全ガイドシリーズ
▶ 各論シリーズ
Vol.1:第1番〜第8番染色体
致死的トリソミーから8番モザイクまで。初期流産の原因を解説
Vol.2:第9番〜第16番染色体
13番パトウ・15番微細欠失・16番流産リスクの核心
▶ 関連する重要記事
参考文献
- [1] Hassold T, Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet. 2001;2(4):280-291. [PubMed]
- [2] Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet. 1991;49(5):995-1013. [PubMed]
- [3] Kaur A, et al. Trisomy 8 mosaicism syndrome. BMJ Case Rep. 2012;2012:bcr2012006281. [PMC]
- [4] Bianchi DW, et al. DNA sequencing versus standard prenatal aneuploidy screening. N Engl J Med. 2014;370(9):799-808. [PubMed]
- [5] Grati FR, et al. Outcomes in pregnancies with a confined placental mosaicism and implications for prenatal screening using cell-free DNA. Genet Med. 2020;22(2):309-316. [PubMed]
- [6] ACMG Board of Directors. Points to consider in the clinical application of genomic sequencing. Genet Med. 2012;14(8):759-761. [ACMG]
- [7] Shaffer LG, et al. Rare autosomal trisomies detected by cell-free DNA screening: a summary of 8 studies. Prenat Diagn. 2017;37(8):725-731. [PubMed]
- [8] Kalousek DK, Vekemans M. Confined placental mosaicism. J Med Genet. 1996;33(7):529-533. [PubMed]
- [9] GeneReviews – 1p36 Deletion Syndrome. University of Washington, Seattle. [GeneReviews]
- [10] Orphanet – Wolf-Hirschhorn Syndrome. [Orphanet]
- [11] Orphanet – Cri du Chat Syndrome. [Orphanet]
- [12] Williams Syndrome Association. Clinical Information. [Williams Syndrome Association]
- [13] ISPD Position Statement. The use of cell-free DNA for prenatal screening for chromosomal abnormalities. Prenat Diagn. 2015;35(8):725-734. [ISPD]