目次

Q. 16トリソミーとは何ですか?流産との関係は?
A. 16トリソミーは、自然流産の原因として最も頻度が高い染色体異常です。
全常染色体トリソミーの中で約30%(流産全体の15〜20%)を占め、完全型は100%致死的です。ただし、モザイク型として一部が出生に至ることがあり、NIPTの全染色体検査で妊娠早期から検出可能です。
-
➤
16トリソミー → 流産原因No.1、全常染色体トリソミーの約30%を占める -
➤
13トリソミー(パトウ症候群) → 出生可能だが予後不良、生存期間中央値7〜10日 -
➤
15番染色体異常 → プラダー・ウィリー/アンジェルマン症候群と関連 -
➤
モザイク型 → 完全型が致死的でもモザイク型として出生する可能性あり -
➤
全染色体検査 → ダイヤモンドプランで13/15/16番を含む6種のトリソミーを検査可能
📚 全染色体検査ガイドシリーズ
- ➤染色体異数性とは(総論)
- ➤Vol.1|第1番〜第8番染色体
- ★Vol.2|第9番〜第16番染色体(この記事)
- ➤Vol.3|第17番〜第22番・性染色体
1. 16トリソミーとは?流産原因No.1の理由
【結論】 16トリソミーはヒトの自然流産において最も頻度の高い染色体異常です。全常染色体トリソミーの約30%、流産全体の15〜20%を占めます。完全型は100%致死的ですが、モザイク型として出生に至るケースもあります。
「流産の原因は何だったのだろう」「また同じことが起きるのではないか」そんな不安を抱えている方も多いでしょう。流産を経験されると、原因を知りたいと思うのは当然のことです。
実は、初期流産(妊娠12週未満)の50〜70%は染色体異常が原因とされています。その中でも16トリソミーは最も多く、流産検体の約7%以上から検出されます。
💡 用語解説:トリソミーとは?
トリソミー(Trisomy)とは、通常2本ずつある染色体が3本になっている状態のことです。16トリソミーは16番染色体が3本ある状態を指します。染色体の過剰は遺伝子の発現バランスを崩し、多くの場合は発生初期に致死的となります。
16トリソミーが流産原因No.1である理由
16トリソミーがこれほど高頻度で発生する理由には、以下の要因が考えられています。
減数分裂エラー
- •
母体年齢の上昇と強く相関
- •
卵母細胞の減数分裂I期でエラーが起きやすい
- •
16番染色体は不分離が起きやすい構造的特徴を持つ
致死性の高さ
- •
完全型は100%致死的
- •
着床後まもなく発生が停止
- •
「空の胎嚢」として観察されることも多い
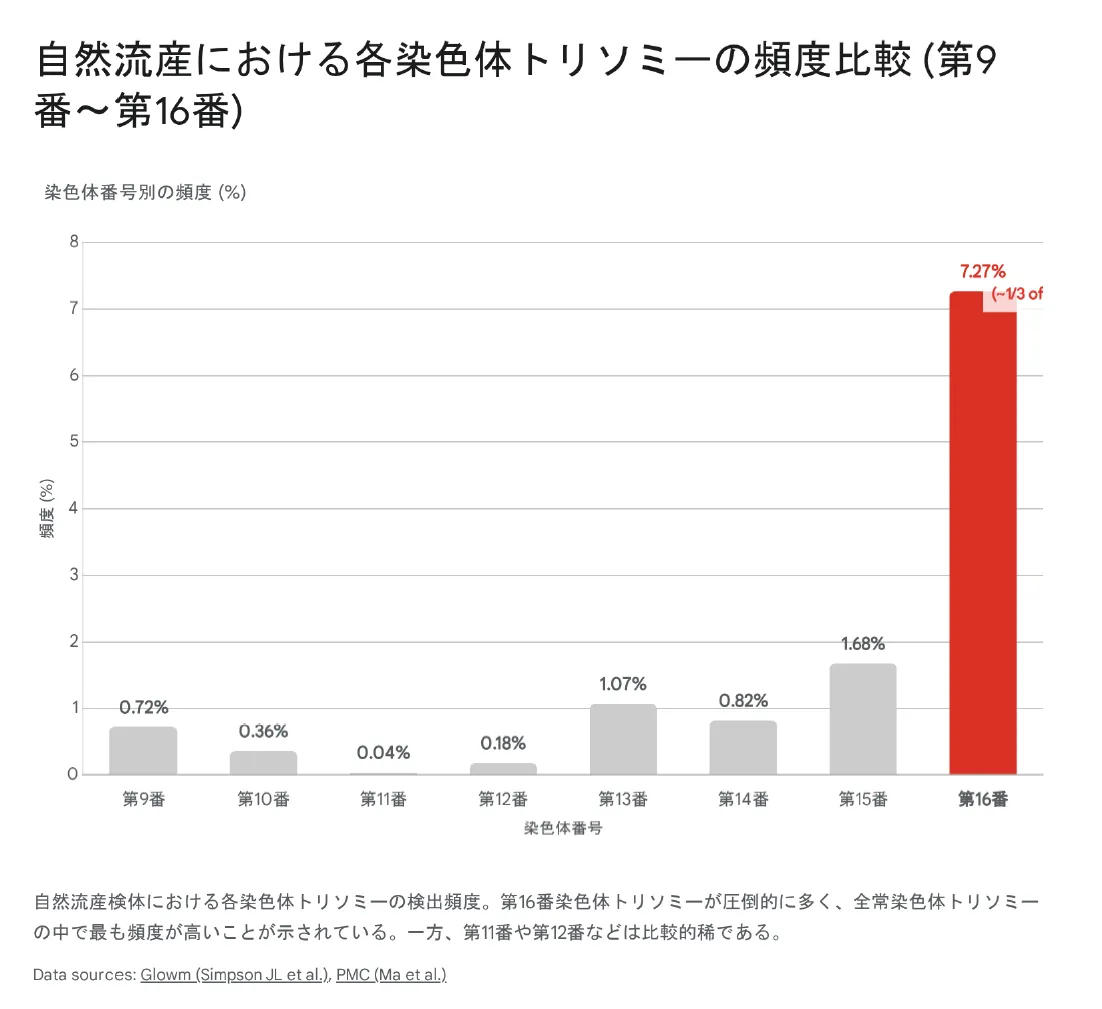
2. 第9番〜第16番染色体トリソミーの頻度比較
【結論】 9〜16番染色体のトリソミーの中で、16トリソミーが圧倒的に多く(約7.27%)、次いで15トリソミー(約1.68%)、13トリソミー(約1.07%)の順です。13番以外は完全型では致死的ですが、モザイク型として出生する可能性があります。
下のグラフは、自然流産検体における各染色体トリソミーの検出頻度を示しています。16番染色体トリソミーが他を大きく引き離して最も高頻度であることがわかります。
▼ 自然流産における各染色体トリソミーの頻度比較(第9番〜第16番)
| 染色体番号 | 流産検体での頻度 | 完全型の予後 | モザイク型での出生 |
|---|---|---|---|
| 第9番 | 約0.72% | 致死的 | 稀(約150例の報告) |
| 第10番 | 約0.36% | 致死的 | 極めて稀(数例のみ) |
| 第11番 | 約0.04% | 致死的 | 極めて稀(3例程度) |
| 第12番 | 約0.18% | 致死的 | 極めて稀(PKSとして) |
| 第13番 | 約1.07% | 出生可能 | パトウ症候群として出生 |
| 第14番 | 約0.82% | 致死的 | 稀(30〜40例) |
| 第15番 | 約1.68% | 致死的 | 極めて稀(PWS/ASと関連) |
| 第16番 | 約7.27% | 致死的 | 比較的多い(CPMとして) |
💡 なぜ13番だけが出生可能なのか?
13番、18番、21番染色体は、遺伝子数が比較的少ない染色体です。そのため、トリソミーになっても発生プログラムが完全には破綻せず、出生に至ることがあります。一方、16番染色体は遺伝子数が平均的であるにもかかわらず流産で最多となっており、減数分裂時の不分離が起きやすい構造的特徴があると考えられています。
3. 各染色体異常の特徴と予後(第9番〜第12番)
【結論】 第9番〜第12番染色体のトリソミーは、完全型では致死的ですが、モザイク型として稀に出生することがあります。特に12番染色体のテトラソミー(パリスター・キリアン症候群)は診断に注意が必要です。
第9番染色体トリソミー(モザイク型)
完全型トリソミー9は致死的ですが、モザイク型として約150例が世界で報告されています。生存期間の中央値は20日未満と報告されており、多くは生後早期に死亡します。
-
•
頭蓋顔面異形:小頭症、深く窪んだ眼窩、「魚のような口」
-
•
骨格異常:関節拘縮、脱臼、脊椎異常
-
•
内臓奇形:先天性心疾患(VSD、ASD)、腎奇形
-
•
予後:適切な管理で1歳超の生存例あり、成人期まで生存する部分トリソミーの報告も
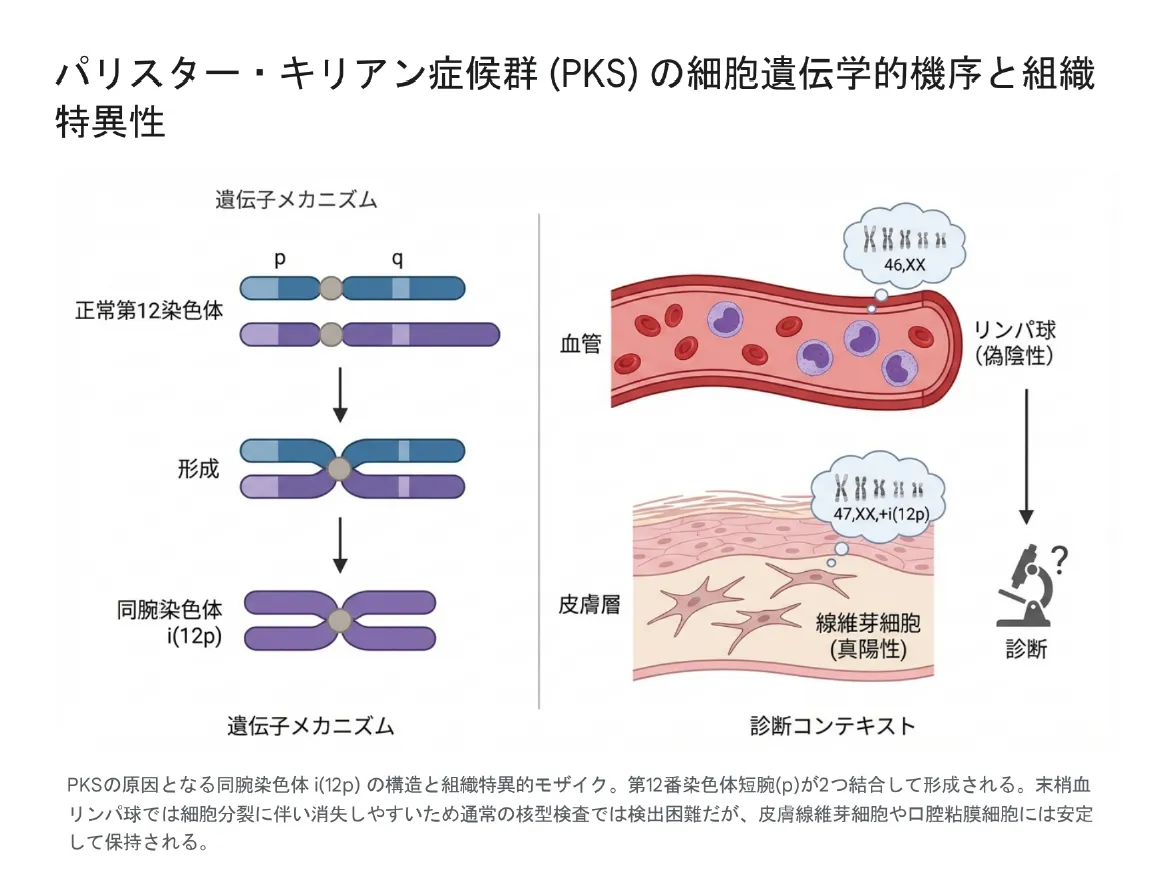
第12番染色体異常:パリスター・キリアン症候群(PKS)
パリスター・キリアン症候群は、12番染色体短腕(12p)のテトラソミー(4コピー)による疾患です。通常の血液検査では検出されにくいという診断上の大きな特徴があります。
⚠️ 診断上の注意点:PKSの「組織限定的モザイク」
PKSの原因となる異常染色体(i(12p))は、血液細胞からは急速に排除される傾向があります。そのため、通常の血液染色体検査では「正常」と誤診されるリスクが高いのです。PKSを疑う場合は、皮膚線維芽細胞や口腔粘膜細胞を用いた検査が推奨されます。
▼ パリスター・キリアン症候群(PKS)の細胞遺伝学的機序と組織特異性

🩺 院長コラム【「見逃されやすい」染色体異常について】
パリスター・キリアン症候群のように、通常の血液検査では見つけにくい染色体異常が存在します。「血液検査で正常だったから大丈夫」と安心してしまうケースもありますが、臨床症状と検査結果が一致しない場合は、別の組織での検査を検討する必要があります。
当院では臨床遺伝専門医として、検査結果の解釈だけでなく、「どの検査をどの組織で行うべきか」という判断まで含めてサポートしています。検査の選択から結果の解釈まで、一貫して専門医が対応することの重要性を、日々の診療で実感しています。
4. 13番染色体異常:パトウ症候群の特徴と予後
【結論】 13トリソミー(パトウ症候群)は、出生頻度約1/10,000〜1/20,000の重篤な染色体異常です。生存期間の中央値は7〜10日ですが、近年は積極的治療により生存期間が延長する傾向にあります。
パトウ症候群は、21トリソミー(ダウン症候群)、18トリソミー(エドワーズ症候群)に次いで3番目に頻度の高い常染色体トリソミーです。当院の13トリソミー専門ページで詳しく解説していますので、ここでは概要をお伝えします。
パトウ症候群の主な特徴
中枢神経系・顔面
- •
全前脳胞症(HPE)
- •
両側性口唇口蓋裂
- •
小眼球症・無眼球症
四肢・皮膚
- •
軸後性多指症(小指側の過剰指)
- •
揺り椅子状足底
- •
頭皮欠損
内臓奇形
- •
先天性心疾患(80%以上)
- •
多発性嚢胞腎
- •
消化管奇形
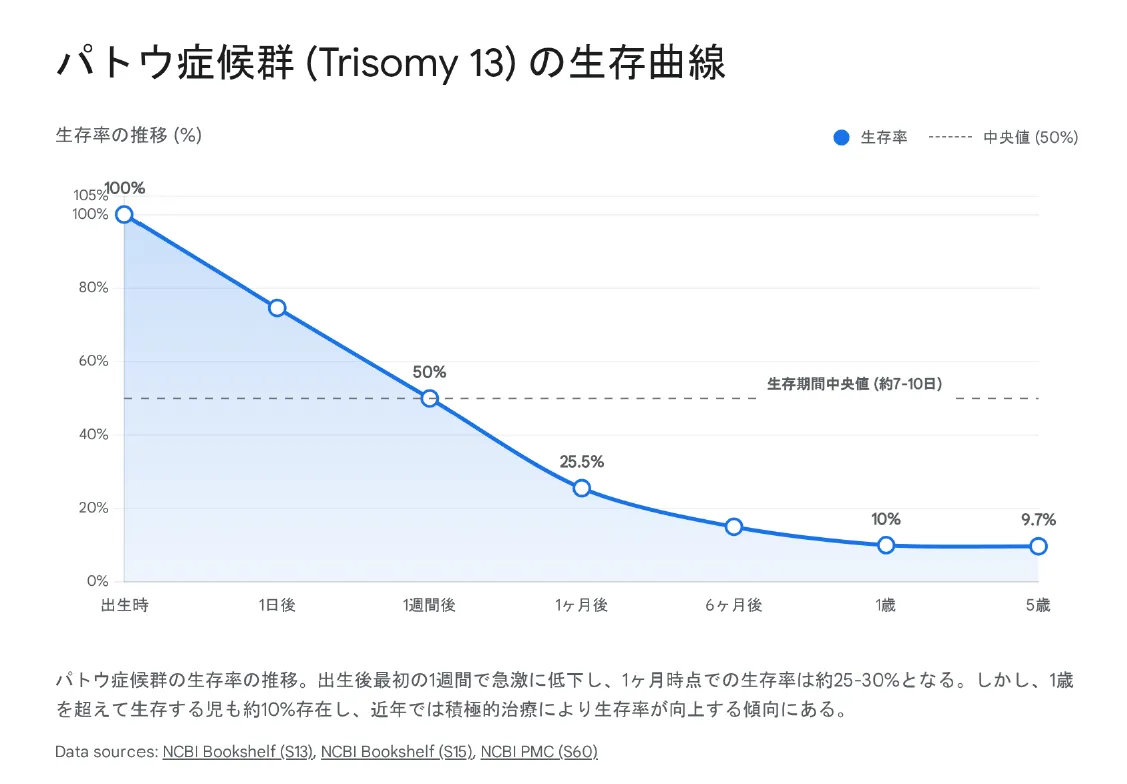
生存率の推移
▼ パトウ症候群(Trisomy 13)の生存曲線
| 時点 | 生存率 | 備考 |
|---|---|---|
| 出生時 | 100% | — |
| 1週間後 | 約50% | 生存期間中央値(7〜10日) |
| 1ヶ月後 | 約25〜30% | — |
| 1歳 | 約10% | 積極治療群では改善傾向 |
| 5歳 | 約9.7% | 10年以上の長期生存例も報告 |
💡 治療方針のパラダイムシフト
かつて「致死的」とされていたパトウ症候群ですが、近年は「生命を脅かす(Life-limiting)」状態として捉え直す動きがあります。心臓手術などの積極的介入を行ったコホートでは、生存期間中央値が733日まで延長したという報告もあります。家族の価値観に基づいた意思決定(Shared Decision Making)が重視されるようになっています。
5. モザイク型と限局性胎盤モザイク(CPM)
【結論】 完全型トリソミーが致死的な染色体でも、モザイク型として出生する可能性があります。特に16トリソミーでは限局性胎盤モザイク(CPM)が比較的多く、胎児は正常でも胎盤機能不全を引き起こすリスクがあります。
💡 用語解説:モザイク現象とは?
モザイク現象(Mosaicism)とは、正常な細胞と異常な細胞が体内に混在している状態のことです。発生初期に一部の細胞で染色体の数が変化すると、その後の細胞分裂で正常細胞と異常細胞が混在するようになります。異常細胞の割合や分布によって、症状の重さは大きく異なります。
16トリソミーの転帰:流産から生存への分岐
16トリソミーの受精卵は、多くが自然流産に至りますが、一部は「トリソミーレスキュー」という救済機構により生存への道が開かれます。
▼ 第16番染色体トリソミーの転帰:流産から生存への分岐
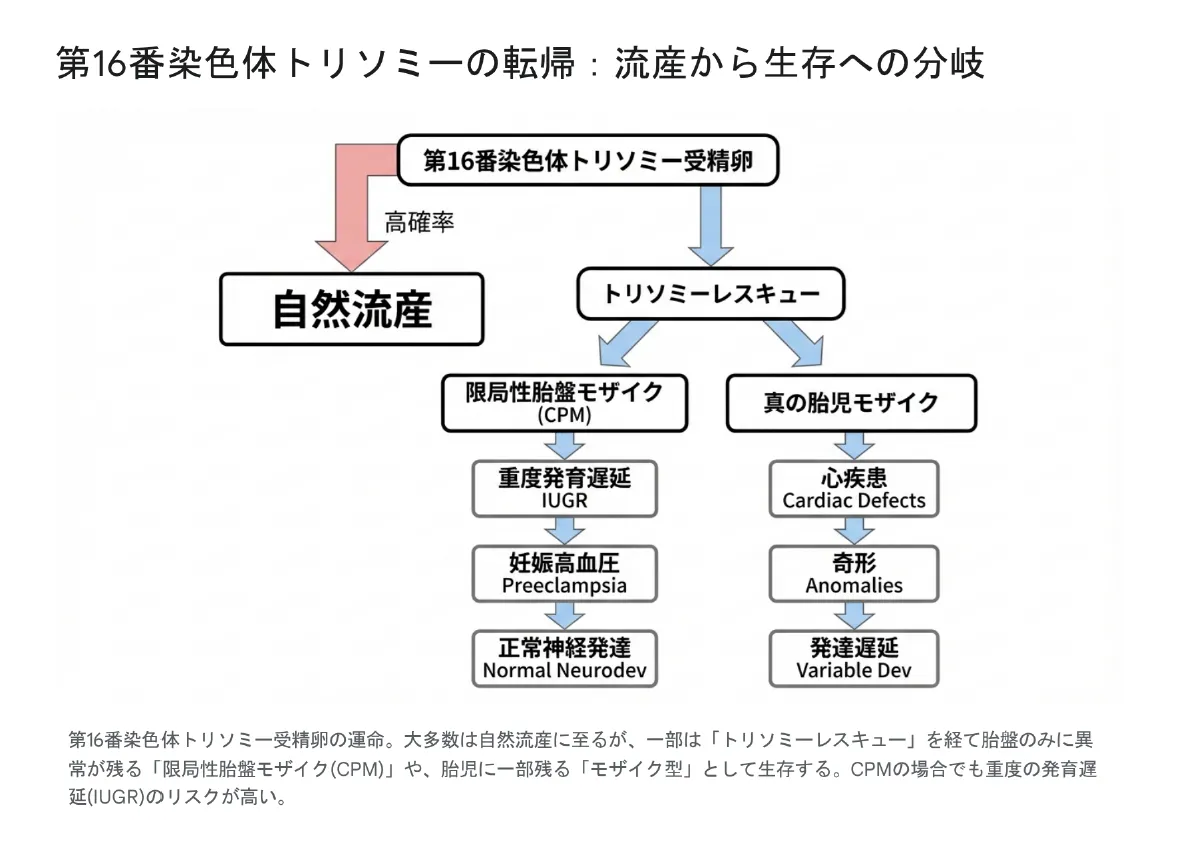
-
①
定義:異常な細胞が胎盤組織のみに限定され、胎児本体は正常核型を持つ状態
-
②
NIPTとの関係:NIPTは胎盤由来のDNAを検出するため、CPMで偽陽性になることがある
-
③
産科的リスク:重度の子宮内発育遅延(IUGR)、妊娠高血圧腎症、早産のリスク上昇
-
④
予後:出生後の神経発達は正常〜軽度遅延にとどまる例が多い、キャッチアップ成長が見られる
14番・15番染色体と片親性ダイソミー(UPD)
14番と15番染色体はインプリンティング遺伝子(親の由来によって発現が異なる遺伝子)を含んでいます。トリソミーレスキューの結果、片親性ダイソミー(UPD)が生じると、特有の症候群を発症するリスクがあります。
| 染色体 | UPDの親由来 | 関連する症候群 | 主な症状 |
|---|---|---|---|
| 第14番 | 母性UPD14 | Temple症候群 | 低身長、早発思春期、小手小足 |
| 父性UPD14 | Kagami-Ogata症候群 | ベル状胸郭、重度呼吸障害(予後不良) | |
| 第15番 | 母性UPD15 | プラダー・ウィリー症候群 | 筋緊張低下、過食・肥満、知的障害 |
| 父性UPD15 | アンジェルマン症候群 | 重度知的障害、発語欠如、失調性歩行 |
染色体異常について不安を感じていませんか?
モザイク型、CPM、UPD…複雑な情報に混乱することも。
臨床遺伝専門医がわかりやすく説明します。
※オンライン診療も対応可能です
🧬 各染色体異常の詳細(第9番〜第16番)
本記事で解説した第9番から第16番染色体のそれぞれの異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクからさらに詳細をご確認いただけます。
6. NIPTで全染色体検査を受けるメリット
【結論】 NIPTの全染色体検査により、13/15/16番を含む複数のトリソミーを妊娠早期から検査できます。特にダイヤモンドプランのCOATE法は陽性的中率99.9%超の高精度を実現しています。
「21/18/13トリソミー以外も検査できるの?」「全染色体検査のメリットは?」そんな疑問をお持ちの方も多いでしょう。当院では、より広範囲の染色体異常を検出できる全染色体検査を提供しています。
ダイヤモンドプラン(COATE法)の検査範囲
🔬 ダイヤモンドプランで検査できる項目
常染色体トリソミー(6種)
13 / 15 / 16 / 18 / 21 / 22
性染色体異数性(4種)
45,X / 47,XXX / 47,XXY / 47,XYY
微細欠失(12領域)
DiGeorge症候群など重要な領域
単一遺伝子(56遺伝子)
症候性自閉症関連を含む30+疾患
詳細はダイヤモンドプラン専用ページをご覧ください
COATE法の精度
| 検査項目 | 従来法の陽性的中率 | COATE法の陽性的中率 |
|---|---|---|
| 微細欠失 | 約70%台 | >99.9% |
| 56遺伝子(新生突然変異) | — | >99.9% |
COATE法のエビデンスについては、こちらの専門ページで詳しく解説しています。
ミネルバクリニックのトリプルリスクヘッジ
💰 金銭的リスクヘッジ
互助会(8,000円)で羊水検査費用を全額カバー(上限なし)。陽性時の経済的不安を解消します。
⏱️ 時間的リスクヘッジ
2025年6月〜院内で羊水検査・絨毛検査が可能に。転院不要で時間を短縮。
🤝 心理的リスクヘッジ
臨床遺伝専門医が最初から最後まで担当。院内完結で患者様のトラウマを防ぎます。
💡 遺伝カウンセリング料金について
当院の検査費用には遺伝カウンセリング料金(33,000円相当)が含まれています。これは当日の説明だけでなく、陽性時に何度でもカウンセリングを受けられる権利、妊娠経過中の心配事(サイトメガロウイルス感染など)への相談も含まれています。「お金がかかるから相談しにくい」ということがないよう配慮しています。
🩺 院長コラム【「正確性」を最優先する理由】
NIPTには「2日で結果が出る」ことを売りにする施設もありますが、当院は「スピード」より「正確性」を最優先しています。
なぜなら、NIPTの結果はお子さんの生涯、そしてご家族の人生に関わる重大な情報だからです。「早く知りたい」というお気持ちはよくわかります。しかし、不正確な結果による誤った判断は、取り返しがつきません。
当院では米国4大遺伝子検査会社の技術を採用し、バイオインフォマティクスの専門家による解析を経て結果をお届けしています。偽陰性ゼロのエビデンスは、この「正確性への徹底したこだわり」の結果です。
よくある質問(FAQ)
🏥 一人で悩まないでください
染色体異常について心配なこと、検査を受けるかどうか迷っていること、
どんなことでもお気軽にご相談ください。
臨床遺伝専門医があなたとご家族に寄り添います。
📚 全染色体検査・完全ガイドシリーズ
▶ 各論シリーズ
Vol.1:第1番〜第8番染色体
致死的トリソミーから8番モザイクまで。初期流産の原因を解説
Vol.2:第9番〜第16番染色体
13番パトウ・15番微細欠失・16番流産リスクの核心
▶ 関連する重要記事
参考文献
- [1] Ma J, et al. Aneuploidy in Early Miscarriage and its Related Factors. Chin Med J (Engl). 2016;129(3):303-7. [PMC]
- [2] Simpson JL. Genetic and Nongenetic Causes of Pregnancy Loss. Global Library of Women’s Medicine. [GLOWM]
- [3] Hassold T, et al. Prenatally Diagnosed Rare Trisomy 16 Mosaicism. Mol Syndromol. 2018;9(4):185-195. [PMC]
- [4] Benn PA. Trisomy 16 and trisomy 16 mosaicism: a review. Am J Med Genet. 1998;79(2):121-33. [PubMed]
- [5] Patel C, et al. Pallister-Killian syndrome: clinical, cytogenetic and molecular findings in 15 cases. Mol Syndromol. 2018;9(5):239-249. [PMC]
- [6] Patau K, et al. Trisomy 13 – StatPearls. NCBI Bookshelf. [StatPearls]
- [7] Carey JC. Health supervision and anticipatory guidance for children with genetic disorders. Pediatr Clin North Am. 1992;39(1):25-53. [PubMed]
- [8] Kotzot D, Utermann G. Uniparental disomy (UPD) other than 15: phenotypes and bibliography updated. Am J Med Genet A. 2005;136(3):287-305. [PubMed]
- [9] Cassidy SB, Driscoll DJ. Prader-Willi syndrome. Eur J Hum Genet. 2009;17(1):3-13. [PMC]
- [10] American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 226: Screening for Fetal Chromosomal Abnormalities. Obstet Gynecol. 2020;136(4):e48-e69. [ACOG]