目次
ソトス症候群とは?
顔つきの特徴・症状・寿命を専門医が解説

ソトス症候群はNSD1遺伝子の変異による過成長症候群です。特徴的な顔つき(顔貌)の年齢による変化から、大人になってからの寿命や自立まで、臨床遺伝専門医が正確な情報と今後の見通しを丁寧に解説します。
Q. ソトス症候群とはどのような病気ですか?
A. 5番染色体のNSD1遺伝子の変異や欠失によって引き起こされる過成長症候群です。
かつて「脳性巨人症」とも呼ばれ、過成長(高身長・大頭症)・特徴的顔貌・知的発達の遅れの3主徴を特徴とします。約95%は親からの遺伝ではなく、新生突然変異として発生します。
-
➤
原因 → NSD1遺伝子のハプロ不全(機能の半減) -
➤
3主徴 → 過成長、特徴的な顔つき、発達の遅れ -
➤
予後・寿命 → 生命予後は良好で、寿命は健常者と変わりません -
➤
診断 → 臨床所見と遺伝子検査(確定診断) -
➤
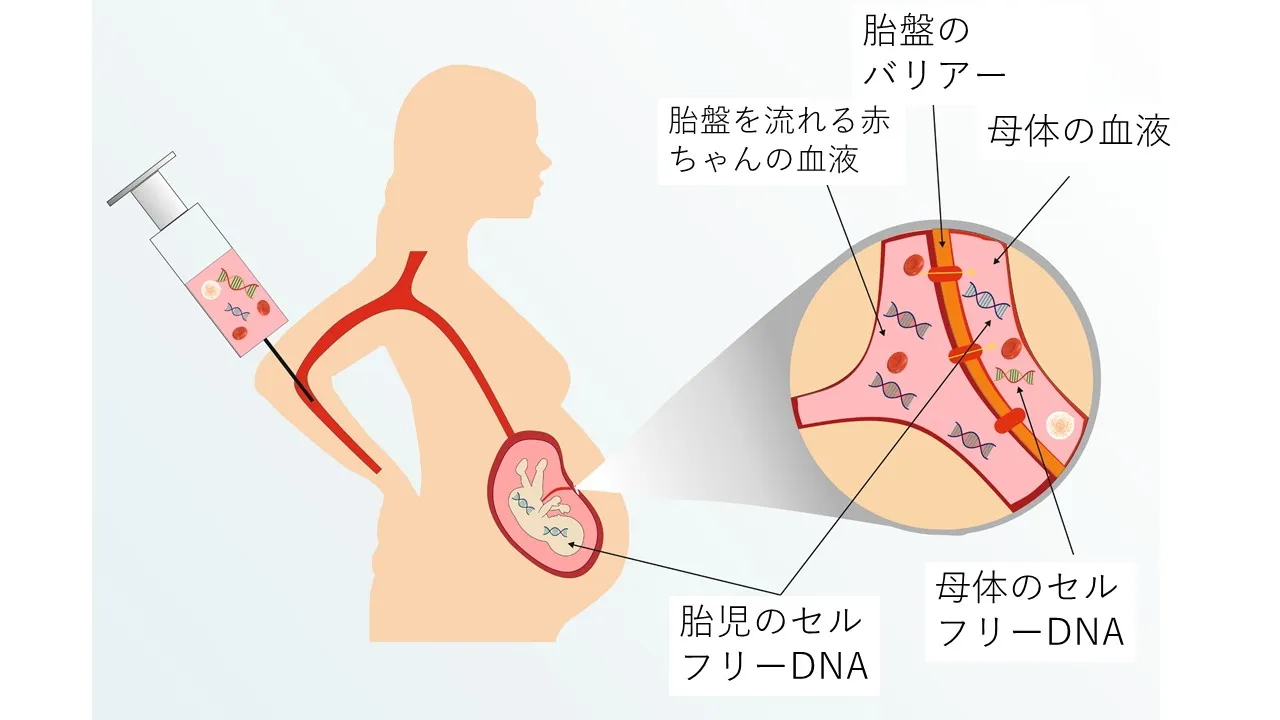
NIPT → 妊娠中の検査としてNIPTでスクリーニングも可能です
1. ソトス症候群とは|基本情報
【結論】 ソトス症候群は、NSD1遺伝子の変異または欠失によって引き起こされる過成長症候群です。1964年にJuan Sotosにより初めて報告され、「脳性巨人症」とも呼ばれました。約1/14,000出生と推定され、最も頻度の高い遺伝性過成長症候群の一つです。
乳幼児期から顕著な高身長と大頭症(巨頭症)を示し、特徴的な顔つき(顔貌)と知的発達の遅れを伴う遺伝性疾患です。2002年に日本の黒滝らによってNSD1遺伝子が原因遺伝子として同定されました。
💡 用語解説:「過成長症候群」とは?
身長や頭囲が同年齢の平均より著しく大きい(通常+2SD以上)ことを特徴とする遺伝性疾患群の総称です。ソトス症候群のほか、Weaver症候群などが含まれます。
ソトス症候群の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | ソトス症候群(Sotos syndrome) |
| 別名 | 脳性巨人症(Cerebral Gigantism)※現在は使用されにくい |
| 原因遺伝子 | NSD1遺伝子(5q35) |
| 遺伝形式 | 常染色体優性(顕性)遺伝 |
| 発生頻度 | 約1/14,000出生 |
| 新生突然変異率 | 約95%(親からの遺伝は稀) |
⚠️ 「脳性巨人症」という名称について
かつて呼ばれていた名称ですが、成長ホルモンの異常ではなく遺伝子の問題であることが判明したため、現在では病態を正確に反映していない古い名称とされています。
2. ソトス症候群の主な症状|3主徴と顔つきの変化
【結論】 ソトス症候群の3主徴は①過成長(高身長・巨頭症)、②特徴的な顔つき(顔貌)、③知的発達の遅れです。
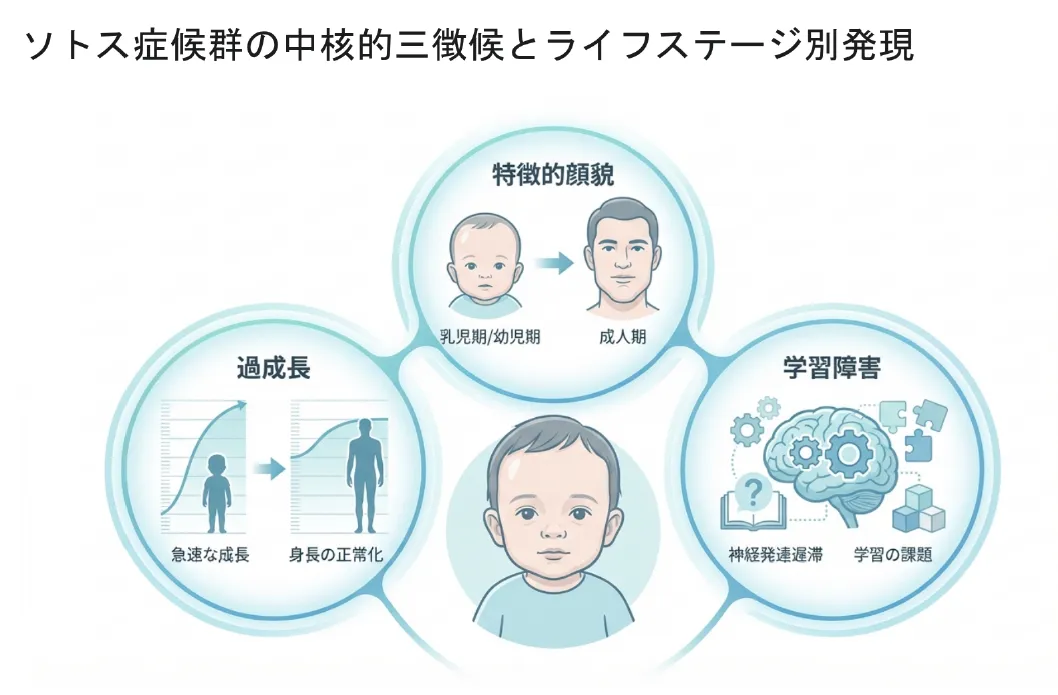
3主徴① 過成長(身体的過成長)
高身長と巨頭症(大頭症)が乳児期から顕著に見られます。小児期には平均より+2SD以上高くなることが多いですが、思春期以降に成長が落ち着き、大人(成人期)の最終身長は正常上限範囲に収まることが大半です。
💡 停留水頭症について
頭部MRIで脳室拡大が認められることが多いですが、これは脳圧亢進を伴わない良性の「停留水頭症」であることが多く、手術を要するケースは稀です。
3主徴② 特徴的な顔つき(年齢による変化)
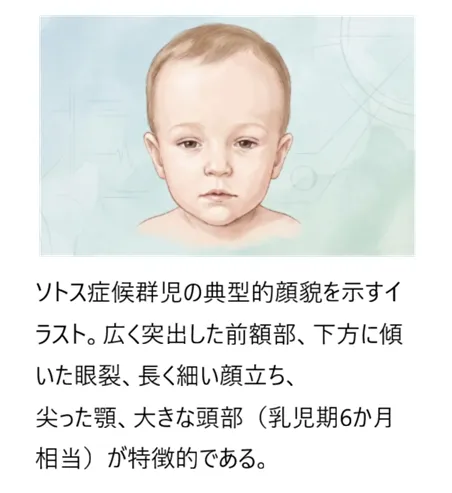
顔面の特徴
- •
広く突出したおでこ(前額部)
- •
眼瞼裂の下方傾斜(下がり目)
- •
長頭傾向(頭の前後が長い)
下顔面の特徴
- •
長く細い顔立ち
- •
尖った顎(幼児期)
- •
紅潮しやすい頬
👶 年齢による「顔つき」の変化
ソトス症候群の特徴的な顔つき(顔貌)は、年齢とともに変化していくのが大きな特徴です。ご家族が「顔つきが気になる」と感じやすい時期と、その後の経過は以下のようになります。
-
▶︎
赤ちゃん〜幼児期(1〜6歳):
最も顔つきの特徴(広いおでこ、下がり目、尖った顎)がはっきりと現れる時期です。この時期の違和感から受診につながるケースが多く見られます。 -
▶︎
学童期:
徐々に顔の長さが目立つようになり、顎がしっかりしてきます。 -
▶︎
大人(成人期):
顔つきの特徴は年齢を重ねるごとにマイルドになり、目立たなくなる傾向があります。尖っていた顎は四角くしっかりとした輪郭へと落ち着きます。
3主徴③ 知的発達の遅れ
乳幼児期から発達の遅れ(首座りや歩行の遅れ、言語の遅れ)がみられ、多くは軽度〜中等度の知的障害が認められます。
- 比較的良好:言語能力、視空間認知・記憶
- 相対的に弱い:非言語的推論、数量的推論
- 改善の可能性:成人期までに発達遅滞が改善し、正常知能に達する人もいます。

3. 原因遺伝子(NSD1)と遺伝的背景
原因は、5番染色体長腕(5q35)に位置するNSD1遺伝子の異常(ハプロ不全)です。
💡 専門用語解説:ヒストンメチルトランスフェラーゼ
NSD1タンパク質はこれに該当します。DNAが巻き付いている「ヒストン」に目印(メチル基)を付ける酵素で、特定の遺伝子のスイッチをON/OFFする重要な働き(エピジェネティクス制御)を持っています。
💡 専門用語解説:ハプロ不全
一対(2つ)ある遺伝子のうち、片方が欠失や変異によって機能しなくなることで、全体として必要なタンパク質の量が半分に減ってしまい、正常な機能が維持できなくなる状態を指します。
⚠️ 日本人の特徴
日本人患者では、遺伝子内変異だけでなく、5q35の微小欠失(ごく小さな染色体の抜け落ち)が約50%以上と多いことが報告されています。
4. 診断方法とNIPT
診断は、3主徴の臨床評価と、NSD1遺伝子検査による確定診断の組み合わせで行います。シークエンス解析や染色体マイクロアレイ(CMA)が用いられます。
また、妊娠中であれば、ミネルバクリニックのNIPT(新型出生前診断)において、NSD1遺伝子を含む56遺伝子パネル検査でのスクリーニングが可能です。
5. 鑑別診断
症状が似ている他の過成長症候群(Weaver症候群、Beckwith-Wiedemann症候群など)との鑑別が重要です。
⚠️ 腫瘍リスクの違い
ソトス症候群では腫瘍リスクの有意な上昇は認められていません。一方、Beckwith-Wiedemann症候群では腫瘍リスクが約15%と高く、定期的なスクリーニングが必要です。
6. 治療と管理
根本的な治療法はありませんが、症状に応じた対症療法や、理学療法(PT)・言語療法(ST)などの早期療育が、お子さんのポテンシャルを最大限に伸ばす鍵となります。てんかんや側弯症への対応も重要です。
7. 大人の寿命と予後(生活の質)
⏳ ソトス症候群の寿命と大人になってからの生活
ご家族から最も多く寄せられるご質問が「寿命は短いのでしょうか?」というものです。結論から申し上げますと、ソトス症候群の方の寿命は、健常者(一般の方)と変わらないと考えられています。生命を直接脅かす合併症がなければ予後は良好です。
知的発達の面でも、年齢とともに落ち着きや理解力が増し、適切な支援環境を整えることで、就労し、社会の一員として自立した生活を送っている大人の方も多数いらっしゃいます。
8. 遺伝カウンセリング
約95%は新生突然変異のため、次子への再発リスクは通常1%未満です。遺伝カウンセリングでは、出生前診断の選択肢など、ご家族が納得のいく決断ができるよう正確な情報を提供します。
よくある質問(FAQ)
関連記事
参考文献
- [1] Tatton-Brown K, et al. Sotos syndrome. Eur J Hum Genet. 2007. [PubMed]
- [2] Kurotaki N, et al. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet. 2002. [PubMed]
- [3] Tatton-Brown K, Rahman N. Sotos Syndrome. GeneReviews. [GeneReviews]
- [4] 難病情報センター. ソトス症候群(指定難病194). [難病情報センター]