目次

メチルマロン酸血症・ホモシステイン血症 cblX型は、X染色体上のHCFC1遺伝子の変異によって引き起こされる、きわめて稀な先天性代謝異常症です。代謝酵素そのものが壊れているのではなく、その酵素の「遺伝子発現スイッチ」を制御する転写調節因子に異常が生じるという、代謝学においてこれまで知られていなかった全く新しいメカニズムで発症します。難治性てんかん・重度知的障害・特徴的な頭蓋顔面奇形を呈し、「リボソーム病」としての側面も併せ持つ複合症候群です。
Q. cblX型とはどんな病気ですか?まず結論だけ知りたいです
A. HCFC1遺伝子の変異によって、コバラミン(ビタミンB12)を活性型に変換するMMACHCタンパク質の産生が低下し、メチルマロン酸とホモシステインが体内に蓄積するX染色体連鎖性の先天性代謝異常症です。男性のみが重症化し、難治性てんかん・重度知的障害・頭蓋顔面奇形を呈します。代謝酵素遺伝子ではなく「転写調節因子」の変異が原因という、代謝学で初めて発見されたメカニズムが特徴です。
- ➤疾患の定義 → OMIM 309541、X連鎖性劣性遺伝、HCFC1遺伝子(Xq28)変異
- ➤分子メカニズム → 転写調節因子HCF-1の機能不全 → MMACHC発現低下 → コバラミン代謝全般の停止
- ➤主な症状 → 難治性てんかん・重度知的障害・頭蓋顔面奇形・MMA蓄積・ホモシステイン蓄積
- ➤鑑別診断 → cblC型・非ケトン性高グリシン血症(NKH)・cblK型との違いを詳解
- ➤治療 → ヒドロキソコバラミン用量強化療法・ベタイン・L-カルニチン。笑気ガスは絶対禁忌
1. cblX型とは:疾患の定義と歴史的背景
メチルマロン酸血症・ホモシステイン血症 cblX型(別名:HCFC1異常症/MAHCX)は、X染色体長腕(Xq28)に位置するHCFC1遺伝子の変異によって引き起こされる稀少な先天性代謝疾患です。体内でビタミンB12(コバラミン)を活性型に変換する際に欠かせない酵素「MMACHCタンパク質」の産生が著しく低下し、代謝産物であるメチルマロン酸(MMA)とホモシステインが全身の組織に有害な濃度で蓄積します。
💡 用語解説:X連鎖性劣性遺伝(エックスれんさせいれっせいいでん)
X染色体上の遺伝子に変異がある場合の遺伝形式です。男性(XY)はX染色体を1本しか持たないため、変異があるX染色体を受け継ぐと正常なコピーがなく発症します(ヘミ接合体)。一方、女性(XX)は2本のX染色体を持つため、もう一方の正常なX染色体が補完的に働き、通常は症状が現れない「保因者(キャリア)」となります。これがcblX型が男性のみで重症化する根本的な理由です。
cblX型は、長年にわたって臨床の場で謎とされてきた問題に答えを与えました。細胞補完群解析でcblC型と同じ所見を示すにもかかわらず、原因遺伝子とされるMMACHC遺伝子には一切変異が見つからない男性患者が散見されていたのです。この謎が解けたのは2013年のことです。米国国立ヒトゲノム研究所(NHGRI)を中心とする国際共同研究チームが、次世代シーケンシングによる全エクソーム解析を用いて、これらの患者の真の原因遺伝子がHCFC1であることを突き止めました。
この発見が臨床遺伝学にもたらしたパラダイムシフトは計り知れません。それまでの先天性代謝異常症は「代謝酵素をコードする遺伝子の欠損」が原因とされてきました。ところがcblX型は、代謝酵素ではなく、その酵素の遺伝子発現を制御する「転写調節因子」の異常によって代謝経路が二次的に破綻するという、それまで観察されたことのない全く新しいメカニズムを持つ疾患でした。この疾患はOMIM(#309541)に「Methylmalonic aciduria and homocysteinemia, cblX type(MAHCX)」として、また「X連鎖性知的発達障害3型(MRX3, XLID3)」としても登録されています。
2. 原因遺伝子HCFC1と分子病態メカニズム
cblX型の複雑な病態を理解するには、正常なコバラミン代謝の流れと、その流れを上位から制御する転写調節ネットワークを理解することが不可欠です。
2-1. 正常なコバラミン代謝:2つの活性型とその役割
💡 用語解説:コバラミン(ビタミンB12)の細胞内処理
食事から摂取したビタミンB12(コバラミン)は、そのままでは酵素の補酵素として機能できません。細胞内に取り込まれた後、MMACHCタンパク質の働きによって2種類の活性型に変換されます。
①アデノシルコバラミン(AdoCbl):ミトコンドリア内の酵素「メチルマロニルCoAムターゼ(MCM)」の補酵素。アミノ酸や脂肪酸の異化に必須。
②メチルコバラミン(MeCbl):細胞質の酵素「メチオニン合成酵素(MS)」の補酵素。有害なホモシステインをメチオニンに変換する「再メチル化」に必須。
MMACHCが機能しなければ、この2経路が同時に停止します。
AdoCbl経路が止まると、バリン・メチオニン・イソロイシン・スレオニンなどのアミノ酸や奇数鎖脂肪酸の分解が滞り、メチルマロニルCoAから変換されるべきサクシニルCoAが作られず、代わりに有毒なメチルマロン酸(MMA)が蓄積します。一方、MeCbl経路が止まると、再メチル化サイクルが機能不全に陥り、血中にホモシステインが異常蓄積し(高ホモシステイン血症)、メチオニンが枯渇します。
2-2. HCFC1遺伝子とHCF-1タンパク質の役割
💡 用語解説:転写共役因子(HCF-1)とは
HCFC1遺伝子がコードするHCF-1(Host Cell Factor-1)タンパク質は、それ自身はDNAに直接結合しません。その代わり、DNAのプロモーター領域(遺伝子発現のスイッチ部分)に結合できる転写因子THAP11(別名RONIN)やZNF143と物理的に結合して複合体を形成し、MMACHCプロモーターに作用して転写を活性化させる「転写共役因子」として機能します。HCF-1なしでは、THAP11もZNF143もMMACHCを十分に発現させることができません。
つまり、HCFC1に変異が生じる → HCF-1の立体構造が乱れる → THAP11・ZNF143との相互作用が失われる → MMACHCプロモーターへの結合ができなくなる → MMACHCの転写が大幅に低下する → コバラミン代謝が全停止する、という連鎖が起きます。
2-3. Kelchドメイン変異とcblX型の発症
cblX型の典型的な臨床像(MMA血症+ホモシステイン血症+重篤な神経症状)を示す患者に最も多く見られるのが、HCF-1タンパク質N末端側の「Kelchドメイン」内のミスセンス変異です。代表的なものとしてGln68・Ala73・Ala115残基への置換(例:p.Ala115Val)が報告されています。これらのアミノ酸置換はタンパク質の高次構造(コンフォメーション)に不可逆的な変化をもたらし、THAP11・ZNF143との正常な相互作用能力を失わせます。
2-4. 二重の病態:代謝異常症+リボソーム病
💡 用語解説:リボソーム病(リボソームパシー)とは
リボソームとは、細胞内でタンパク質を合成する工場のような構造体です。リボソームの構築(リボソーム生合成)が先天的に障害されると、細胞の増殖・分化・器官形成が広汎に影響を受けます。HCF-1とそのパートナーRONIN(THAP11)は、MMACHCの転写調節だけでなく、胚発生期における「リボソーム生合成」の重要な転写調節因子としても機能していることが、遺伝子改変マウスを用いた研究で明らかになっています。
HCFC1変異マウスの細胞(MEF)を解析すると、AdoCbl・MeCblの低下に加えて、リボソームタンパク質のサブユニットをコードする多数の遺伝子群の発現が有意に低下していることが確認されました。これはリボソームの構築そのものが根本的に阻害されていることを意味します。
この知見こそが、cblX型の病態を理解する上での核心です。cblX型は単なる「コバラミン代謝異常症」ではなく、「重篤な細胞内コバラミン代謝異常症」と「リボソーム病」の2つの病態が同時に生じる複合症候群です。cblC型では見られないような頭蓋顔面奇形や脳の構造的形成異常がcblX型で著明に現れるのは、このリボソーム生合成の破綻と細胞周期・細胞分化の乱れが組み合わさっているためです。
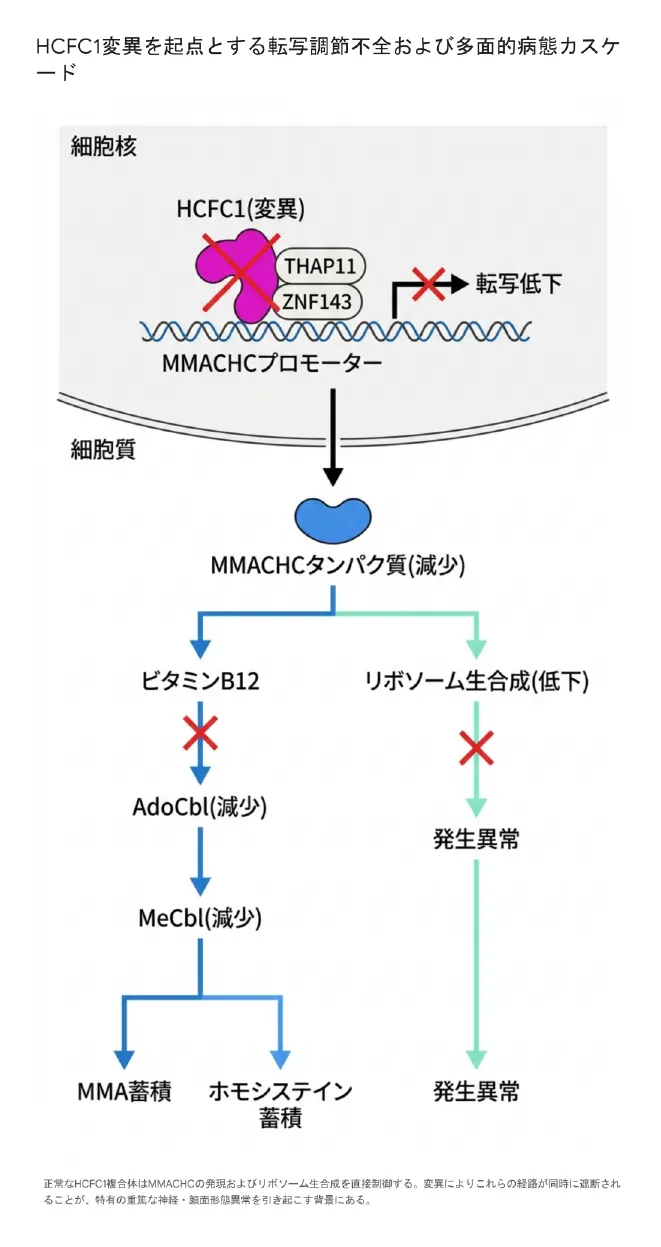
HCFC1変異を起点とする転写調節不全の病態カスケード。THAP11・ZNF143との複合体が機能を失い、MMACHCの転写低下とリボソーム生合成障害が同時に引き起こされる。
3. 主な症状と臨床像
cblX型はX連鎖性劣性遺伝であるため、臨床的に重篤な症状を呈するのは罹患したX染色体を持つ男性(ヘミ接合体)に限られます。発症は極めて早期で、胎児期の子宮内発育遅延から生後数か月の乳児期に集中します。その重症度は同じ複合型コバラミン代謝異常症であるcblC型を大きく凌駕します。
💡 用語解説:ヘミ接合体(hemizygote)とは
通常、私たちは各染色体を2本ずつ持っています。しかし男性のX染色体は1本のみです。X染色体上の遺伝子に変異がある場合、男性には「正常なコピー」がなく、変異のある遺伝子の影響がそのまま現れます。この状態をヘミ接合体と呼びます。cblX型は、X染色体を1本しか持たない男性だけが重症化する理由がここにあります。
3-1. 中枢神経系への影響:難治性てんかんと知的障害
cblX型で最も特筆すべき臨床的特徴は、難治性てんかん(intractable epilepsy)の高頻度な合併です。乳児期早期から始まる重度の筋緊張低下(哺乳力の著しい低下)を皮切りに、点頭てんかん(乳児スパスム)・強直発作・間代発作、さらにミオクロニー発作へと多様な発作型が年齢とともに変遷します。
これらの発作は既存の多くの抗てんかん薬に対して強い抵抗性(難治性)を示し、コントロールが極めて困難です。同時に、生後早期から重度の精神運動発達遅滞が顕在化し、知的発達障害へと至ります。脳の構造的形成異常(brain malformations)・小頭症(microcephaly)・舞踏アテトーゼなどの錐体外路系不随意運動も頻繁に報告されています。
MRI(神経画像検査)では、内包後脚から始まり白質全体へと広がる拡散制限やADC(見かけの拡散係数)値の低下など、脳内高グリシン血症に関連した特異的な白質障害所見が観察されることがあります。これは後述するNKH類似の生化学プロファイルとも関連します。
3-2. 頭蓋顔面・骨格系の奇形
HCFC1の変異は、胚発生段階における神経堤細胞(neural crest cells)の増殖能と分化に直接的な影響を与えます。神経堤細胞は頭蓋顔面領域の骨・軟骨・結合組織の形成に不可欠な多能性前駆細胞であり、その機能不全は独特の外見的特徴をもたらします。
🧠 神経系・発達
- 重度精神運動発達遅滞・知的障害
- 難治性てんかん(多様な発作型)
- 重度筋緊張低下(hypotonia)
- 小頭症
- 舞踏アテトーゼ(錐体外路症状)
- 脳の構造的形成異常
👤 頭蓋顔面・骨格
- 長頭(elongated face)
- 大きな耳介
- クモ状指(arachnodactyly)
- 細長い体型(lean body habitus)
- 顔面の異形症(facial dysmorphia)
- 顔面の非対称性
🔬 生化学的異常
- ↑ 血中・尿中・CSF中MMA
- ↑ 血漿総ホモシステイン(tHcy)
- ↓ 血清メチオニン
- ↑ CSF中グリシン(NKH模倣)
- ↑ プロピオニルカルニチン(C3)
- ↑ メチルクエン酸、3-OH-プロピオン酸
🩸 全身・血液
- 巨赤芽球性貧血・血球減少症
- 哺乳不良・成長障害
- 異常な蒼白(pallor)
- 子宮内発育遅延(IUGR)
- 色素性網膜症は通常みられない
(cblC型との差異)
モデル生物のゼブラフィッシュを用いたノックダウン実験(hcfc1b遺伝子の抑制)でも、メッケル軟骨・角舌軟骨・全鰓弓軟骨の完全な消失という深刻な頭蓋顔面の構造異常が確認されており、ヒト患者の臨床像と一致します。これらの形態学的発達異常は、単なるMMACHCの機能不全(cblC型)では生じない特徴であり、HCF-1によるリボソーム生合成と細胞増殖の制御不全という二重の病態によって引き起こされます。
3-3. 重要な生化学的落とし穴:NKHへの誤診リスク
💡 用語解説:非ケトン性高グリシン血症(NKH)とは
グリシン開裂酵素系の欠損によって脳脊髄液(CSF)中にグリシンが異常蓄積する先天性アミノ酸代謝異常症です。新生児期からの重篤なてんかんと知的障害を特徴とし、CSFと血漿のグリシン比が上昇することが診断の手がかりとなります。cblX型でも同様のCSFグリシン上昇が見られることがあり、初期診断でNKHと誤診されるリスクがあります。
cblX型の患者では、CSF中グリシン濃度が異常に上昇し、髄液/血漿グリシン比が上昇することがあります。このプロファイルはNKHの古典的な生化学的所見を模倣(mimic)するため、診断初期段階でNKHと誤診される危険性が高く、臨床医の注意が必要です。cblX型の疑いがある場合は、必ずMMA・ホモシステイン・メチオニンを同時測定することが重要です。

4. 鑑別診断:他のコバラミン代謝異常症との違い
cblX型の確定診断において最大の課題は、同様の生化学的プロファイル(MMA上昇+ホモシステイン上昇)を呈する他の遺伝性代謝疾患との鑑別です。特に、最初の新生児スクリーニング結果だけでは各病型を区別することができません。
| 疾患(病型) | 原因遺伝子 | 遺伝形式 | MMA | tHcy | 鑑別のポイント |
|---|---|---|---|---|---|
| cblX型(本疾患) | HCFC1 | X連鎖性劣性 | ↑↑ | ↑↑ | 男性のみ発症。難治性てんかん・頭蓋顔面奇形・リボソーム病。CSFグリシン上昇。MMACHC陰性後にHCFC1を検索 |
| cblC型 | MMACHC | 常染色体劣性 | ↑↑ | ↑↑ | 最多(全体の80%)。色素性網膜症(bull’s eye maculopathy)が特徴的。乳児期HUSリスク。男女とも発症 |
| cblJ型 | ABCD4(ATP結合カセットサブファミリーD) | 常染色体劣性 | ↑ | ↑ | リソソームからのコバラミン輸送障害。男女とも発症。遺伝子検査で鑑別 |
| cblF型 | LMBRD1 | 常染色体劣性 | ↑ | ↑ | リソソームコバラミン輸送障害。男女とも発症。遺伝子パネルで区別 |
| cblD型 | MMADHC | 常染色体劣性 | ↑ | ↑ | 変異部位によりMMAのみ・Hcyのみ・両方の3亜型あり。男女とも発症 |
| 孤立性MMA(cblB型等) | MMAB等(関連:ACADM) | 常染色体劣性 | ↑↑ | 正常 | ホモシステインが上昇しない点でcblX型と明確に区別可能。代謝性アシドーシス・高アンモニア血症が主体 |
| CBS欠損症 | CBS | 常染色体劣性 | 正常 | ↑↑ | メチオニンが著明に上昇する点でcblX型(メチオニン低下)と正反対。水晶体脱臼・マルファン様骨格 |
| NKH(非ケトン性高グリシン血症) | GLDC等 | 常染色体劣性 | 正常 | 正常 | CSFグリシン上昇のみ。MMAもtHcyも上昇しない点でcblX型と区別できる。cblX型のNKH模倣に注意 |
| cblK型(THAP11異常症) | THAP11 | 常染色体劣性 | ↑ | ↑ | cblX型と類似の表現型。男女とも発症(常染色体劣性)。脳奇形・難治性てんかん。下記参照 |
cblX関連疾患群(cblX様疾患):THAP11異常症とZNF143異常症
cblX型の発見は、HCF-1と複合体を形成する他の転写因子群の変異による新たな類縁疾患の同定へとつながりました。これらは「cblX様疾患(cblX-like disorder)」または「cblK型」として総称されています。
THAP11異常症(RONIN)
常染色体劣性遺伝。HCF-1と直接相互作用し、MMACHCプロモーターに結合する転写因子の変異。代表変異:c.240C>G, p.Phe80Leu。
臨床:生後2か月からの難治性てんかん、中等度MMA血症、重度知的障害。最大限の治療介入にもかかわらず10歳で死亡した報告あり。男女とも発症。
ZNF143異常症(cblK型)
HCF-1と協調するZNF143の複合ヘテロ接合体変異。MMACHCの転写低下に加え、特異な細胞内現象を伴う。
特徴:リソソーム内にトランスコバラミン結合コバラミンが異常蓄積。AdoCbl合成:正常の3.4%、MeCbl合成:正常の1.0%まで激減。既存の補完群とも異なる独特の代謝欠陥。
5. 診断・遺伝子検査の進め方
cblX型の確定診断には段階的なアプローチが必要です。新生児スクリーニング(NBS)から始まり、生化学的検査、そして分子遺伝学的解析へと進みます。
5-1. 新生児マス・スクリーニング(NBS)の役割と限界
タンデム質量分析計(LC-MS/MS)を用いた新生児乾燥濾紙血(DBS)スクリーニングでは、cblX型を含む複合型コバラミン代謝異常症がプロピオニルカルニチン(C3)の上昇・メチオニンの低下・C3/C0比の異常などとして検出される可能性があります。近年はDBSからメチルクエン酸やホモシステインを直接検出する技術も進歩しています。
しかしながら、NBSの生化学的所見だけでcblX型を他のコバラミン代謝異常症(cblC・cblD・cblF・cblJ型など)から区別することは不可能です。確定診断には必ず分子遺伝学的アプローチが必要です。
5-2. 確定診断:全エクソーム解析(WES)がゴールドスタンダード
💡 用語解説:全エクソーム解析(WES)とは
WES(Whole Exome Sequencing)とは、ゲノムのうちタンパク質をコードする領域(エクソン)全体を網羅的に解析する次世代シーケンス技術です。全遺伝子を一度に調べることができるため、原因遺伝子が特定されていない場合や複数の可能性がある場合に特に有効です。cblX型のように「まずMMACHC遺伝子が陰性で、次に何を調べるか」という状況でこそ力を発揮します。クリニカル・エクソーム解析とも呼ばれます。
推奨される確定診断への流れは以下の通りです。
🔬 cblX型 確定診断フロー
- NBS・血漿MMA・tHcy・メチオニン・尿中有機酸などの生化学的スクリーニング実施
- 複合型コバラミン代謝異常症を疑う → MMACHC遺伝子の単独または遺伝子パネル検査(メチルマロン酸尿症・ホモシスチン尿症NGSパネル)
- MMACHC陰性 → 速やかに全エクソーム解析(WES)またはクリニカル・エクソーム解析へ移行(核・ミトコンドリアNGS遺伝子検査)
- X染色体上HCFC1遺伝子にヘミ接合体(男性)または複合ヘテロ接合体(稀に女性での関与)の病的バリアントを同定 → 確定診断
X連鎖性遺伝形式の確認、そしてKelchドメイン(Xq28)内のミスセンス変異であること(非症候性知的障害を引き起こす他のドメイン変異との区別)が確定診断の要件です。コバラミン・ホモシステイン・メチオニンの測定に特化した遺伝子検査については、コバラミン・ホモシステイン・メチオニン遺伝子検査のページもご参照ください。
6. 治療・長期管理プロトコル
現時点では、HCFC1遺伝子の変異そのものを修復する根本的な治療法は臨床実装されていません。治療の主軸は急性期の代謝危機の安定化と、生化学的マーカーの長期コントロールを目的とした多面的な内科的・栄養学的管理です。
6-1. ヒドロキソコバラミン用量強化療法:治療の根幹
💡 用語解説:ヒドロキソコバラミンとは
ビタミンB12(コバラミン)の活性前駆体の1種です。経口のシアノコバラミン製剤とは異なり、注射で直接血中に投与することで細胞内に高濃度で到達させることができます。細胞内で欠乏している活性型(AdoCblとMeCbl)の前駆物質として機能し、障害された代謝経路をある程度補完します。cblX型の治療において最も根幹をなす薬剤です。
近年の臨床研究では、早期発症型患者に対してヒドロキソコバラミンを5・25・50 mg/mLという高濃度で調製し、毎日の静脈内(IV)または皮下(SQ)注射で持続投与する「用量強化(Dose Intensification)」戦略の有効性が報告されています。この治療により、
- ✓血漿総ホモシステイン(tHcy)の顕著な低下
- ✓尿中・血中MMA(uMMA, MMA)の持続的改善
- ✓CSF-MMAおよび髄液グリシン濃度の低下
- ✓枯渇していたメチオニン濃度の回復、一部症例では難治性てんかんの発作頻度が減少
が確認されています。頻回の注射によるQOL低下を防ぐため、皮下注射用ポート(SQ injection port)の外科的留置が推奨されます。ただし、早期から治療を開始し生存率が改善されても、HCF-1異常に起因するリボソーム機能不全や胚発生段階での神経構築異常を後天的に補正することは科学的に不可能であり、進行性の認知・神経障害や頭蓋顔面奇形を完全に予防・回復させることは極めて困難という臨床的限界を認識しておく必要があります。
6-2. 補助療法
💡 用語解説:ベタインとは
肝臓に存在する代替的なメチル基供与体。ベタイン-ホモシステインメチルトランスフェラーゼ(BHMT)という別の酵素を介して、ホモシステインをメチオニンへ再メチル化することができます。メチオニン合成酵素(MS)が機能不全に陥っているcblX型において、ホモシステインの血中濃度を下げ、メチオニンを補充する迂回路として機能します。経口投与が可能です。
ヒドロキソコバラミンに加え、以下の補助療法が組み合わされます。ベタイン(無水ベタイン)の経口投与:再メチル化の代替経路を活性化し、ホモシステイン低下とメチオニン補充に寄与します。L-カルニチンの補充:有毒な有機酸(プロピオニルCoAなど)が蓄積すると細胞内の遊離カルニチンが消費・枯渇し、ミトコンドリアのエネルギー産生が打撃を受けます。毒性代謝物をアシルカルニチン抱合体として尿中に排泄させるためにL-カルニチンの継続投与が推奨されます。
6-3. 急性代謝不全(急性期)の集中管理と透析
急性期には直ちに全てのタンパク質摂取を停止し、グルコース等による高カロリー輸液(静脈内投与)を開始して身体を同化状態へ強制的にシフトさせ、異化を抑制することが救命の第一歩です。内科的介入でも毒性代謝物の制御ができない極重篤例では、速やかに腎代替療法(透析)が適応されます。血液透析(HD)はMMAクリアランスの効率が最も高い一方、乳幼児では技術的制約から腹膜透析(PD)が選択されることもあります。透析液は重炭酸ベース(bicarbonate-based)を優先使用することがガイドラインで強く推奨されています(乳酸・酢酸ベースはアシドーシスを悪化させるリスクあり)。
6-4. 絶対禁忌:笑気ガス(亜酸化窒素)とメチオニン制限
🚨 絶対禁忌:笑気ガス(亜酸化窒素、N₂O)
歯科治療や麻酔導入時に用いられる笑気ガスは、cblX型患者に対して絶対的禁忌(absolute contraindication)です。亜酸化窒素はコバラミン分子の中心のコバルトイオンを酸化し、メチオニン合成酵素を不可逆的に不活化します。吸入により急激かつ致命的な代謝不全、または不可逆的な亜急性連合性脊髄変性症を引き起こす危険性が極めて高いです。必ず医療者・歯科医師に伝えてください。
また、食事療法における重大な誤りとして、cblX型に「メチオニン制限食」を適用してはなりません。CBS欠損症などではホモシステインを下げる目的でメチオニン制限が行われますが、cblX型の患者では再メチル化障害により既にメチオニンが枯渇状態にあります。人為的なメチオニン制限は致死的なタンパク質合成不全・脳の髄鞘形成不全を招きます。栄養管理の基本は、患者の成長に必要な推奨タンパク質許容量を維持しつつ過剰摂取を避ける繊細なバランスの維持にあります。経口摂取が困難な例では早期の胃瘻(ガストロストミー)造設が予後改善に寄与します。
7. 遺伝カウンセリングの意義
cblX型の確定診断後は、患者本人と家族への丁寧な遺伝カウンセリングが不可欠です。X連鎖性劣性遺伝という形式を正しく理解し、家族計画や将来の支援について情報をもとに意思決定できるよう支援します。
- ➤遺伝形式と再発リスクの説明:母親が保因者(ヘテロ接合体)の場合、息子(男性)が発症する確率は50%、娘が保因者になる確率は50%です。父親がcblX型であっても、息子には影響がなく(Y染色体を受け継ぐため)、娘は必ず保因者になります。保因者の女性は通常X不活性化により無症候ですが、キャリアスクリーニングによって変異を持つかどうかを知ることができます。
- ➤出生前診断の選択肢:次子を望む場合、既知の変異を対象とした出生前遺伝子診断が選択肢として存在します。絨毛検査や羊水検査により胎児のHCFC1遺伝子を調べることが可能です。
- ➤女性保因者へのケア:保因者の女性は通常無症候ですが、まれにX不活性化パターンの偏りによって軽度の症状が現れることがあります。定期的なフォローアップが推奨される場合があります。キャリアスクリーニングの詳細は米国人類遺伝学会の推奨内容もご参照ください。
- ➤長期的サポート体制の構築:疾患の希少性から国内の患者情報が限られています。重度の知的障害や難治性てんかんを持つお子さんへの長期的な支援体制(療育・福祉・教育)の検討を、医療チームと連携しながら進めることが重要です。
- ➤X連鎖性疾患の遺伝カウンセリング体験談:同じX連鎖性疾患の保因者検査を受けられた方の体験談が参考になります。副腎白質ジストロフィー保因者検査の体験談や、X連鎖性疾患と家族計画(ALD)のコラムも合わせてご覧ください。また、コバラミン代謝に関わる酸化ストレス関連遺伝子としてPRDX1遺伝子の情報も遺伝子一覧でご確認いただけます。
8. よくある誤解
誤解①「cblX型はcblC型と同じ病気」
生化学的プロファイルは類似していますが、cblX型はcblC型より重篤です。難治性てんかん・頭蓋顔面奇形・リボソーム病という側面はcblC型にはなく、発症機序も根本的に異なります。またMMACHC遺伝子検査が陰性という点も重要な違いです。
誤解②「MMACHC陰性ならコバラミン代謝異常ではない」
MMACHC遺伝子が陰性でも、その転写調節因子であるHCFC1の変異によってMMACHCの産生が低下し、まったく同様の代謝異常が生じます。「MMACHC陰性=除外」とはなりません。WESへの移行が必要です。
誤解③「髄液グリシンが高いからNKHだ」
cblX型でも髄液グリシンが上昇するため、NKHと誤診されることがあります。男性乳児でMMAとtHcyが同時に上昇しているならNKHではありません。鑑別のためMMA・tHcy・メチオニンの同時測定が必須です。
誤解④「女性は全く関係ない病気」
重症化するのは男性ですが、女性は保因者として変異を次世代に伝えます。また保因者女性であっても、X不活性化パターンの偏りで軽度症状が現れる場合があります。家族の女性への遺伝カウンセリングも重要です。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 コバラミン代謝異常症・遺伝性代謝疾患のご相談
cblX型をはじめとする先天性代謝異常症・希少遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
参考文献
- [1] Gérard Morin et al. An X-Linked Cobalamin Disorder Caused by Mutations in Transcriptional Coregulator HCFC1. Am J Hum Genet. 2013;93(3):506-514. [PMC3769968]
- [2] MedlinePlus Genetics. Methylmalonic acidemia with homocystinuria. [MedlinePlus]
- [3] NHGRI. NHGRI researchers help identify new metabolic disorder caused by faulty gene expression. 2013. [NHGRI]
- [4] UniProt. Methylmalonic aciduria and homocystinuria, cblX type. DI-03561. [UniProt]
- [5] Tuite A et al. The role of HCFC1 in syndromic and non-syndromic intellectual disability. Medical Research Archives. 2021. [PMC8218923]
- [6] Quintana AM et al. Mutations in Hcfc1 and Ronin result in an inborn error of cobalamin metabolism and ribosomopathy. Hum Mol Genet. 2022. [PubMed 35013307]
- [7] Yu HC et al. Mutations in THAP11 cause an inborn error of cobalamin metabolism and developmental abnormalities. Hum Mol Genet. 2018. [PMC5886234]
- [8] Inborn Error of Cobalamin Metabolism Associated with the Intracellular Accumulation of Transcobalamin-Bound Cobalamin and Mutations in ZNF143. [ResearchGate]
- [9] Quintana AM et al. Hcfc1b, a zebrafish ortholog of HCFC1, regulates craniofacial development by modulating mmachc expression. Dev Biol. 2015. [PMC4391465]
- [10] GeneReviews. Disorders of Intracellular Cobalamin Metabolism. NCBI Bookshelf. [NBK1328]
- [11] Guéant JL et al. Inherited defects of cobalamin metabolism. ResearchGate. 2022. [ResearchGate]
- [12] Parini R et al. Parenteral hydroxocobalamin dose intensification in five patients with different types of early onset intracellular cobalamin defects. JIMD Rep. 2019. [PMC6718108]
- [13] Servais A et al. Clinical Practice Recommendations on Kidney Management in Methylmalonic Acidemia. ERKNet/MetabERN Expert Consensus. 2024. [ERKNet]
- [14] Hcfc1 loss-of-function mutations disrupt neuronal and neural progenitor cells of the developing brain. Hum Mol Genet. 2015. [Oxford Academic]






