目次

📍 クイックナビゲーション
ホモシスチン尿症を伴うメチルマロン酸血症は、ビタミンB12(コバラミン)の細胞内代謝経路における先天的な遺伝子異常によって引き起こされる、極めて稀で重篤な遺伝性代謝疾患です。最も多いcblC型をはじめ複数のサブタイプが存在し、ミトコンドリアと細胞質の双方で代謝が同時にブロックされるという、単独のメチルマロン酸血症(MMA)や単独のホモシスチン尿症とは根本的に異なる複合的な病態が、神経・眼・血液・心臓・腎臓など全身の多臓器に深刻な影響を及ぼします。
Q. ホモシスチン尿症を伴うメチルマロン酸血症とはどのような病気ですか?まず結論から知りたいです
A. ビタミンB12(コバラミン)の細胞内代謝に関わる先天的な遺伝子異常により、血中にメチルマロン酸(MMA)とホモシステインが同時に蓄積する希少遺伝性代謝疾患です。cblC型が最多で全体の約80%を占め、神経・眼・血液・心臓・腎臓など多臓器に重篤な影響をもたらします。市販のビタミンB12サプリでは効果がなく、専門的な治療が不可欠です。
- ➤疾患の定義 → Orphanet登録疾患。cblC型が最多(世界550例以上)。発症率は地域差あり最大37,000人に1人
- ➤病態メカニズム → AdoCblとMeCblの同時欠乏でMMA蓄積・高ホモシステイン・低メチオニンが連鎖して発生
- ➤相補性群の比較 → cblC・cblD・cblF・cblJ・cblXの5群:原因遺伝子と表現型の違いを整理
- ➤特徴的な合併症 → ブルズアイ黄斑症・巨赤芽球性貧血・神経退行・肺動脈高血圧・溶血性尿毒症症候群(HUS)
- ➤診断の鍵 → NBSで「低メチオニン血症」が複合型を単独MMAから区別する決定的な二次マーカー
- ➤治療の鉄則 → ヒドロキソコバラミン筋注必須(シアノコバラミン経口は無効)+ベタイン。タンパク質制限は禁忌
1. 疾患の定義と生化学的概要
ホモシスチン尿症を伴うメチルマロン酸血症(Combined methylmalonic acidemia and homocystinuria)は、ビタミンB12(コバラミン)の細胞内代謝経路における先天的な遺伝子異常によって引き起こされる、希少かつ重篤な遺伝性代謝疾患です。本疾患の最大の特徴は、2種類の活性型コバラミン補酵素の合成が同時に障害されるという複合的な代謝ブロックにあります。単独のメチルマロン酸血症や単独のホモシスチン尿症(シスタチオニンβシンターゼ欠損症など)の臨床像を単純に足し合わせたものとは全く異なる、複雑な全身性障害が引き起こされます。
💡 用語解説:メチルマロン酸(MMA)が蓄積する理由
プロピオニルCoAというエネルギー代謝の中間産物は、メチルマロニルCoAを経て最終的にサクシニルCoAへと変換されます。この変換を行う酵素がメチルマロニルCoAムターゼ(MMUT)です。MMUTが正常に機能するためにはアデノシルコバラミン(AdoCbl)という活性型補酵素が不可欠で、AdoCblが欠乏するとこの変換が止まりメチルマロン酸が血中・尿中に異常蓄積します。本疾患ではミトコンドリア内でのAdoCbl合成が先天的に障害されているため、常にこの代謝ブロックが生じています。
💡 用語解説:ホモシスチン尿症とはなぜ同時に起きるのか
ホモシステインはメチオニンの代謝中間産物です。通常は細胞質内でメチオニンシンターゼ(MTR)という酵素によってメチオニンへと再変換(再メチル化)されますが、MTRの補酵素としてメチルコバラミン(MeCbl)が必須です。本疾患では細胞質内でのMeCbl合成も同時に障害されるため、ホモシステインが蓄積・尿中排泄され、メチオニンが著しく枯渇します。高ホモシステイン血症は血管障害・血栓形成・神経障害を引き起こします。
つまり本疾患では、ミトコンドリア内のAdoCbl不足(→MMA蓄積)と細胞質内のMeCbl不足(→ホモシステイン蓄積・メチオニン枯渇)が、単一の遺伝子異常によって同時に発生します。この「複合的欠陥」こそが本疾患の本質であり、神経・眼・血液・心臓・腎臓を同時に巻き込む複雑な全身性病態の根源となっています。
💡 用語解説:常染色体劣性遺伝とは
本疾患のほとんどのサブタイプ(cblC・cblD・cblF・cblJ型)は常染色体劣性遺伝形式をとります。「常染色体」は性染色体(X・Y)以外の染色体を指し、「劣性」は両親からそれぞれ1つずつ変異遺伝子を受け継いだとき(つまり2コピーの変異を持つとき)に初めて発症することを意味します。両親はそれぞれ変異遺伝子を1つ持つ「保因者(キャリア)」であり、通常は無症状です。両親が保因者の場合、子が発症する確率は理論上25%です。
疫学:cblC型の発症率と地域差
最多のcblC型の発症率は当初「新生児20万人に1人」と推計されていましたが、タンデム質量分析計(MS/MS)を用いた新生児マススクリーニング(NBS)の普及によりこの数字を大幅に上回ることが判明しています。米国ニューヨーク州では約10万人に1人、カリフォルニア州のヒスパニック系集団では約37,000人に1人という極めて高い罹患率が報告されており、世界全体でcblC型だけで550例以上の症例が集積されています。一方でcblD・cblF・cblJ型はそれぞれ世界報告数が20例未満という超稀少疾患です。
2. 病態生理と細胞内コバラミン代謝メカニズム
本疾患の病態を正確に理解するには、細胞内でコバラミンがどのように処理されるかを把握することが鍵です。以下の図はその全体像を示しています。
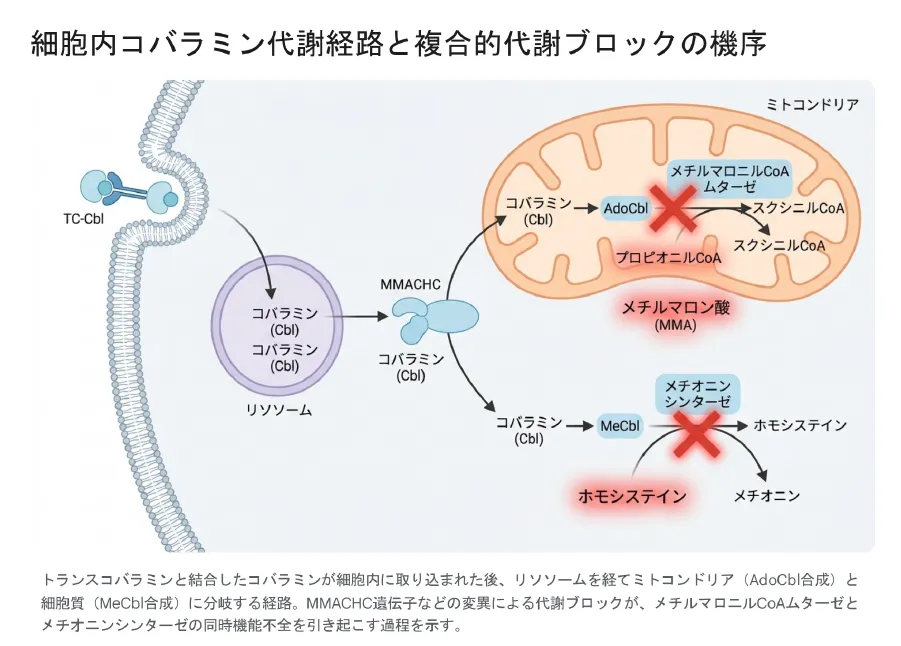
トランスコバラミンと結合したコバラミンが細胞内に取り込まれた後、リソソームを経てミトコンドリア(AdoCbl合成)と細胞質(MeCbl合成)に分岐する経路。MMACHC遺伝子などの変異による代謝ブロックが、メチルマロニルCoAムターゼとメチオニンシンターゼの同時機能不全を引き起こす過程を示す。
食事から摂取されたコバラミンは血中でトランスコバラミン(TC)と結合し、細胞膜上の受容体に認識されてエンドサイトーシスで細胞内に取り込まれます。その後リソソーム内でTCが分解されて遊離したコバラミンが放出され、細胞質へ出ます。このリソソームからの排出過程に異常をきたすのがcblF型(LMBRD1遺伝子)・cblJ型(ABCD4遺伝子)です。
💡 用語解説:MMACHCタンパク質とは(cblC型の核心)
細胞質に放出されたコバラミンは、まずMMACHCタンパク質(MMACHC遺伝子がコード)の働きによって結合基が除去されます。MMACHCはコバラミンをミトコンドリア経路(AdoCbl合成)と細胞質経路(MeCbl合成)に送り出す「振り分け役」として機能する中心的なシャペロン様タンパク質です。cblC型ではMMACHCの機能不全により、AdoCblとMeCblの両方の合成が同時に止まります。これが単独MMAや単独ホモシスチン尿症と全く異なるcblC型特有の複合病態の根源です。
なお、胎児期における病態形成のメカニズムとして重要な仮説があります。胎児組織ではメチオニンシンターゼ(MTR)の基礎活性が成体組織より高く設定されており、正常な器官発生には極めて高いメチル化能力が必要とされます。つまり胎生期のMeCbl依存性再メチル化の阻害が、神経堤細胞や心筋細胞の発生に決定的な悪影響を与え、cblC型に特異な先天性心筋症(左室緻密化障害)や胎児水腫の発症につながると考えられています。

3. 遺伝学的背景と相補性群の比較
本疾患は歴史的に、患者由来の培養線維芽細胞を用いた細胞融合実験による相補性群(Complementation classes)として分類されてきました。現在では各相補性群が特定の遺伝子の異常に起因することが判明しています。
💡 用語解説:相補性群とは
患者の皮膚から採取した培養細胞どうしを人工的に融合させたとき、どの組み合わせで代謝機能が「補完(回復)」されるかを調べることで、異なる遺伝子が原因である患者グループを分類する方法です。同じ相補性群に属する患者は同一の遺伝子に変異があることを意味します。
🔵 cblC型(最多:全体の約80%)
- 責任遺伝子:MMACHC(1p36.3)
- 遺伝形式:常染色体劣性
- 世界報告数:550例以上
- 特徴:早期/遅発型の2型。重篤な神経学的退行・ブルズアイ黄斑症・特異な脳形態異常を伴う複合型の代表
🟣 cblD型
- 責任遺伝子:MMADHC(2q23.2)
- 遺伝形式:常染色体劣性
- 世界報告数:17例(複合型6例)
- 特徴:変異部位により「複合型」「単独MMA」「単独ホモシスチン尿症」の3表現型に分かれる特異な群
🟢 cblF型
- 責任遺伝子:LMBRD1(6q13)
- 遺伝形式:常染色体劣性
- 世界報告数:15例
- 特徴:リソソームからのコバラミン排出障害。哺乳不良・重度筋緊張低下・口内炎・顔貌異常・皮疹
🟡 cblJ型
- 責任遺伝子:ABCD4
- 遺伝形式:常染色体劣性
- 世界報告数:3例程度
- 特徴:cblFと類似したリソソーム排出障害の表現型。先天性心奇形を伴うことがある超稀少群
🔴 cblX型
- 責任遺伝子:HCFC1(X染色体)
- 遺伝形式:X連鎖劣性(男児のみ)
- 特徴:生化学的にはcblC類似のプロファイルを示すが、重度難治性てんかんや顔面形態異常を伴う。MMACHC遺伝子の転写調節の障害が原因
なお、細胞内コバラミン代謝異常にはAdoCbl合成のみが障害される群(cblA・cblB型など)も存在し、これらは「単独のメチルマロン酸血症」を引き起こします。またMeCbl合成のみが障害される群(cblE・cblG型など)は「単独のホモシスチン尿症」を引き起こします。これらは臨床的アプローチや治療方針が根本的に異なるため、本稿が対象とする複合型とは区別が必要です。単独MMAについてはcblB型ビタミンB12反応性MMAのページもご参照ください。
4. 発症時期別の主な症状
本疾患の臨床症状は胎児・周産期から成人期発症(遅発型)まで極めて広いスペクトラムを持ち、発症年齢によって主な障害臓器や症状が大きく異なります。
胎児・周産期の症状
⚠️ 非免疫性胎児水腫・IUGR
明らかな原因が特定できない胎児水腫・羊水過多・子宮内胎児発育遅延(IUGR)・出生時からの小頭症が報告されています。
❤️ 先天性心筋症(cblC型特有)
左室緻密化障害(LVNC)や進行性の拡張型心筋症という特異な先天性心筋症を合併することが確認されており、これが胎児期の難治性心不全や胎児水腫の直接的な原因になります。
💡 用語解説:左室緻密化障害(LVNC)
心臓の左心室の内壁が海綿状(スポンジ状)のままで「緻密化」されない先天性の心筋症です。胎生期に胎児の心臓は内壁が海綿状ですが、通常は妊娠中に滑らかな筋肉に変化します。この変化が起きないのがLVNCで、心不全・不整脈・血栓形成を引き起こし予後不良となります。cblC型のMeCbl依存性代謝ブロックが心筋細胞の正常な発生を妨げると考えられています。
乳幼児期(早期発症型)の症状
生後数週間〜数ヶ月以内に発症する早期発症型は病状の急速な悪化と多臓器不全を特徴とし、未治療の場合の致死率は極めて高くなります。
🧠 神経症状
著明な筋緊張低下・難治性痙攣・全般的発達遅滞・知的障害・進行性小頭症。MRI所見では大脳白質異常・大脳基底核病変・脳梁萎縮が高頻度に確認されます。
🩸 血液学的異常
巨赤芽球性貧血が特徴的で、患児は著明な蒼白を呈します。骨髄での造血障害により汎血球減少症・好中球減少を合併することも稀ではありません。
🫀 腎臓・肺血管
溶血性尿毒症症候群(HUS)・非典型的腎不全(TMA)を発症。肺動脈高血圧(PAH)を合併する症例群も報告されており、発症した場合の予後は極めて不良です。
👁️ 眼科:ブルズアイ黄斑症
cblC型に特異的なブルズアイ黄斑症(牛の目状の黄斑変性)。眼球の浮動運動・眼振・斜視で発見されることが多く、多くが生後10年以内に法的盲に至ります。
💡 用語解説:ブルズアイ黄斑症(Bull’s eye maculopathy)
眼底検査で確認される特徴的な所見で、低色素性の中心部を過色素性の輪が取り囲む「牛の目」のような外観を示す黄斑(網膜の中心)の変性です。cblC型では生後数週間から始まり進行性に悪化します。重要な事実として、全身の生化学値が治療で正常化されても網膜変性の進行を阻止できないことが臨床上の最大の課題です。これはヒドロキソコバラミンが血液網膜関門を十分に通過できない可能性を示唆しています。
💡 用語解説:溶血性尿毒症症候群(HUS)
微小血管の障害によって生じる「溶血性貧血(赤血球が壊れる)+血小板減少+急性腎障害」の三徴を特徴とする重篤な疾患です。本疾患では高ホモシステインが血管内皮細胞に直接的な酸化ストレスを与え、微小血管の血栓形成を促すことで血栓性微小血管症(TMA)が発症し、腎障害に至ります。
学童・成人期(遅発型)の症状
幼児期以降または思春期〜成人期に初めて発症する遅発型は、血液学的異常が軽微であることが多い一方で、特有の重篤な神経・精神症状を呈します。小児期までは正常に発達していた運動・認知機能の急激な退行、学業・職場での成績低下、発話困難、記憶障害から始まり、進行すると認知症様症状・幻覚・精神病様状態(Psychosis)が現れます。
また神経病理学的特徴として、亜急性連合性脊髄変性症(後索・側索の脱髄性変化)による下肢のしびれ・進行性筋力低下・歩行困難・膀胱直腸障害が生じます。さらに再発性の深部静脈血栓症・肺塞栓症などの致死的な血栓塞栓イベントのリスクが成人期を通じて高い点も遅発型管理上の重大な注意事項です。
💡 用語解説:亜急性連合性脊髄変性症
脊髄の後索(感覚に関わる経路)と側索(運動に関わる経路)が同時に傷害(脱髄)される病態で、ビタミンB12欠乏で古典的に知られていますが、本疾患の遅発型でも同じ機序で生じます。感覚・運動の両方が障害されるため、「しびれ(感覚)+筋力低下(運動)」が同時に現れるのが特徴です。
5. 診断と新生児マススクリーニング(NBS)
本疾患における中枢神経系への不可逆的なダメージを防ぐ最大の鍵は早期発見にあり、その最前線を担うのがタンデム質量分析計(MS/MS)を用いた新生児マススクリーニング(NBS)です。
💡 用語解説:タンデム質量分析計(MS/MS)とは
新生児のかかとから採取した血液(乾燥濾紙血・DBS)を分析し、アミノ酸・有機酸・アシルカルニチンなど数十種類の代謝産物を同時に測定できる機器です。わずか数マイクロリットルの血液から多くの先天性代謝異常症を一度にスクリーニングできる現代の新生児スクリーニングの基盤技術です。
スクリーニングアルゴリズム:低メチオニンが鍵
新生児スクリーニングでは、メチルマロン酸血症の一次マーカーとしてプロピオニルカルニチン(C3)の上昇が用いられます。しかしC3の上昇は単独MMAやプロピオン酸血症でも同様に見られるため、これだけでは複合型と区別できません。複合型MMA/ホモシスチン尿症を特定するための決定的な二次マーカーがメチオニン値の低下です。
新生児スクリーニングにおける複合型MMA/HCの鑑別アルゴリズム
単独メチルマロン酸血症(MMA)
または
プロピオン酸血症(PA)
(例:<13.4 µmol/L)
複合型メチルマロン酸血症・ホモシスチン尿症
(cblC, cblD, cblF など)
新生児マススクリーニングにおける乾燥濾紙血(DBS)の一次・二次マーカー解析フロー。C3の上昇に加え、低メチオニン血症の存在がcblC・cblD・cblFなどの再メチル化障害を特定するための重要な分岐点となる。
スクリーニングでcblC型と確定診断された患者の大多数において、NBS時のメチオニン値が13.4 µmol/L未満という低値を示していたことが後方視的解析で明らかになっています。これは「古典型ホモシスチン尿症(CBS欠損症)」でメチオニンが上昇することとは真逆の反応であり、再メチル化障害の特徴的所見です。
確定診断の手順
スクリーニング陽性または臨床的疑いがある場合、以下の組み合わせで確定診断が行われます。
- ➤生化学検査:血漿総ホモシステイン(tHcy)の著明な高値+血清・尿中MMAの顕著な高値+血漿メチオニンの低値
- ➤血清ビタミンB12値が正常範囲内であることを必ず確認(後天性のB12欠乏と区別するため)
- ➤分子遺伝学的検査:コバラミン関連遺伝子検査・NGSパネル検査・または核・ミトコンドリアDNA NGS遺伝子検査によりMMACHC・MMADHC・LMBRD1・ABCD4・HCFC1などの原因遺伝子の両アレル性病原性バリアントを同定
⚠️ 鑑別診断の落とし穴:後天的なビタミンB12欠乏(母体の厳格な菜食主義、悪性貧血、回腸切除による吸収不良など)も全く同一の生化学的プロファイル(MMA↑・tHcy↑)を引き起こします。血清B12値が低値であれば後天性欠乏を先に疑う必要があり、これらを確実に除外してから本疾患を診断します。
6. 治療と長期管理
本疾患には現時点で根治療法は存在しません。しかし適切な薬物療法により酵素機能を部分的に回復させ、有害な代謝産物の蓄積を抑え、枯渇した必須代謝物を補充することで、生存率とQOLを劇的に改善できます。2017年発表のHuemerらによる国際コンセンサスガイドラインが治療の世界標準となっています。
第一選択:ヒドロキソコバラミン(OHCbl)の非経口投与
🚨 重要:シアノコバラミン経口投与は無効です
一般的なサプリメントや経口B12製剤のほとんどはシアノコバラミン(CNCbl)です。本疾患の代謝ブロックを乗り越えることができないため治療効果がありません。治療には必ずヒドロキソコバラミン(OHCbl)の筋肉内(IM)・皮下(SQ)・または静脈内(IV)投与が必要です。症状がある、または再メチル化障害が疑われる患者には、確定診断を待たずにOHCblの投与を速やかに開始することがガイドラインで強く推奨されています。
通常、初期投与量は1日1mg(1000µg)のIM投与から開始し(小児は体重換算0.3 mg/kg/日)、血漿ビタミンB12濃度を1,000,000 pg/ml以上という薬理学的超高値に維持することで、変異によって機能が低下した細胞内酵素システムへ強制的に基質を供給します。
標準的な多剤併用レジメン
💉 ヒドロキソコバラミン(OHCbl)
投与経路:IM/SQ/IV(非経口必須)
用量:0.3 mg/kg/日または1mg/日
血漿B12を超高値に保ち、MMAとtHcyを低下させ、メチオニンと血液学的異常の正常化を図る必須薬剤
💊 無水ベタイン(Betaine)
投与経路:経口
用量:250 mg/kg/日(3回分割)
コバラミン非依存性の代替経路でホモシステインをメチオニンへ再メチル化する極めて重要な補助療法
💊 ホリナートカルシウム/葉酸
用量:5〜15 mg/日
中枢神経系における葉酸欠乏の是正とメチオニン・葉酸サイクルの回転補助
💊 L-カルニチン(Levocarnitine)
用量:50〜100 mg/kg/日(3回分割)
ミトコンドリア内に蓄積した毒性プロピオニルCoAをプロピオニルカルニチンに変換して安全に尿中排泄させ、二次的なカルニチン枯渇を防ぐ
⚠️ タンパク質制限は禁忌:単独MMAとの決定的な違い
単独のメチルマロン酸血症やプロピオン酸血症では、毒性代謝物の産生を抑えるために特定アミノ酸(イソロイシン・バリン・スレオニン・メチオニンなど)のタンパク質制限が標準治療です。しかし複合型MMA・ホモシスチン尿症(cblC等)の患者にタンパク質やメチオニンの厳格な制限を行うことは強く禁忌に近い扱いを受けます。
胎内治療(出生前治療)の実績
過去に罹患児を出産した家族など、胎児がリスクを持つと事前に判明している場合、出生前診断を経て母体を介した胎内治療が選択できます。医学文献では、妊娠15週目から母親に週30mgという超高用量のOHCbl筋注と1日5mgの葉酸経口投与を開始したところ、生まれた子どもが11歳時点でIQ正常・眼科的異常もごく軽微に留まったという画期的な成功例が報告されています。一方で出生後からしか治療を受けられなかった姉(同じcblC型)は重度の知的障害と深刻な視覚障害を持つという対照的な結果となっており、胎生期という極めて早期からの高用量治療が予後を根本から変え得ることが証明されています。
7. 遺伝カウンセリングの意義と家族計画
本疾患の確定診断後、ご本人・ご家族への丁寧な遺伝カウンセリングが不可欠です。
- ➤再発リスクの説明:cblC・cblD・cblF・cblJ型は常染色体劣性遺伝のため、両親がいずれも保因者の場合、次子の発症確率は理論上25%。cblX型はX連鎖劣性で男児のみが発症します。
- ➤保因者(キャリア)検査:ご家族が保因者かどうかを調べることができます。キャリアスクリーニングとは何かをご参照ください。
- ➤出生前診断の選択肢:次子を望む場合、遺伝子検査や羊水検査・絨毛検査による出生前診断が選択肢となります。胎内治療の実施も検討できます。
- ➤X連鎖疾患(cblX)の場合:X連鎖疾患と家族計画の考え方も参考になります。
- ➤多学際的チーム医療:臨床遺伝専門医・小児代謝科医・眼科医・循環器科医・腎臓内科医・神経内科医が連携する体制が生涯にわたり必要です。
8. よくある誤解
誤解①「市販のB12を飲めば治る」
市販のシアノコバラミンや経口製剤では代謝ブロックを越えられず無効です。ヒドロキソコバラミンの筋注という特定の形態での非経口投与が必須です。
誤解②「タンパク質制限が必要」
単独MMAやプロピオン酸血症の管理法を誤って適用しないことが重要です。本疾患でのタンパク質・メチオニン制限は神経退行を悪化させる危険な誤りです。
誤解③「B12が正常だからB12異常ではない」
血清B12値が正常でも、細胞内での代謝が障害されているのが本疾患の本質です。B12値が正常であることが本疾患を否定する根拠にはなりません。
誤解④「治療で血液値が正常化すれば網膜は安全」
cblC型のブルズアイ黄斑症は全身の生化学値が完全に正常化されても進行が止まりません。定期的な眼科専門医受診が生涯を通じて必要です。
9. 予後と臨床遺伝専門医からのメッセージ
新生児スクリーニングの普及とOHCblを中心としたプロトコルの確立により、急性期の致死率は過去数十年で劇的に低下しました。未治療またはシアノコバラミンのみで治療された患者群では全体で30%に達していた死亡率が、NBSによる早期発見によって大幅に改善されています。一方で長期的な罹患率(Morbidity)の改善は依然として大きな課題です。早期発症型の多くは神経認知機能の障害と進行性の網膜変性を抱えたまま成長し、現在の治療でこれを完全に防ぐことは難しいのが現状です。対照的に遅発型では、適切なOHCblとベタイン治療の導入によって認知症様症状・精神症状・脊髄変性が部分的あるいは完全に回復し、社会復帰できた症例が多数報告されています。
🏥 ホモシスチン尿症を伴うメチルマロン酸血症に関するご相談
cblC型をはじめとする細胞内コバラミン代謝異常症の診断・遺伝カウンセリング・
遺伝子検査については、臨床遺伝専門医が在籍するミネルバクリニックにご相談ください。
よくある質問(FAQ)
参考文献・ガイドライン
- [1] Martinelli D, et al. Combined methylmalonic acidemia and homocystinuria, cblC type. I. Clinical presentations, diagnosis and management. J Inherit Metab Dis. 2011;34(6):1275-1283. [PMC4219318]
- [2] Orphanet. Methylmalonic acidemia with homocystinuria. [Orphanet ORPHA:26]
- [3] Froese DS, et al. Disorders of Intracellular Cobalamin Metabolism. In: GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. [NCBI NBK1328]
- [4] Watkins D, Rosenblatt DS. Isolated Methylmalonic Acidemia. In: GeneReviews® [Internet]. [NCBI NBK1231]
- [5] Methylmalonic acidemia with homocystinuria. MedlinePlus Genetics. [MedlinePlus]
- [6] Fischer S, et al. Combined methylmalonic acidemia and homocystinuria, cblC type. II. Complications, pathophysiology, and outcomes. J Inherit Metab Dis. 2012;35(1):103-114. [PMC3529128]
- [7] Huemer M, et al. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J Inherit Metab Dis. 2017;40(1):21-48. [PMC5203859]
- [8] Tsai AC, et al. Newborn screening and early biochemical follow-up in combined methylmalonic aciduria and homocystinuria, cblC type, and utility of methionine as a secondary screening analyte. Mol Genet Metab. 2010;99(2):116-123. [PMC2914534]
- [9] Golzio C, et al. Noncompaction of the Ventricular Myocardium and Hydrops Fetalis in Cobalamin C Disease. JIMD Rep. 2013;7:53-57. [PMC3755569]
- [10] Mütze U, et al. Successful intrauterine treatment of a patient with cobalamin C defect. Mol Genet Metab Rep. 2016;6:55-59. [PMC4789385]
- [11] Kose E, et al. Hemolytic Uremic Syndrome Due to Methylmalonic Acidemia and Homocystinuria in an Infant. Children (Basel). 2021;8(2):112. [MDPI]
- [12] Ricci C, et al. Pilot Study on Neonatal Screening for Methylmalonic Acidemia and Homocystinuria. Metabolites. 2021;7(3):39. [MDPI]






