目次

細胞がビタミンB12(コバラミン)を消化管から吸収しても、その旅はまだ終わりではありません。栄養素はいったん細胞内の「廃棄物処理工場」であるリソソームに閉じ込められ、専用の「扉」を持つタンパク質によって、初めて細胞が使える場所に解放されます。その扉の役割を担うのが、ABCD4遺伝子がコードするリソソーム膜トランスポーターです。長年「誤った場所にいる」と思われ続けていたこのタンパク質の真の姿と、異常が引き起こす重篤な先天性代謝疾患について、臨床遺伝専門医が詳しく解説します。
Q. ABCD4遺伝子とはどのような遺伝子ですか?まず結論だけ知りたいです
A. ABCD4は第14番染色体(14q24.3)にある遺伝子で、リソソーム膜上でビタミンB12(コバラミン)をATPエネルギーを使って細胞質へ運び出すトランスポーターをコードしています。この遺伝子の両方のコピーに機能喪失型変異が起きると、「cblJ型メチルマロン酸血症およびホモシスチン尿症」という重篤な先天性代謝疾患を引き起こします。
- ➤遺伝子の概要 → 14q24.3・19エクソン・606アミノ酸・ABCDサブファミリー・リソソーム膜
- ➤パラダイムシフト → 長年ペルオキシソームタンパクと誤認→実際はリソソームに局在することが判明
- ➤輸送機構 → LMBD1との複合体でリソソームへ輸送され、コバラミンをATP依存的に細胞質へ排出
- ➤関連疾患 → cblJ型メチルマロン酸血症・ホモシスチン尿症(常染色体劣性遺伝・世界で数十例)
- ➤治療の要点 → ヒドロキソコバラミン非経口投与が第一選択。低タンパク食・亜酸化窒素は絶対禁忌
1. ABCD4遺伝子の基本情報
ABCD4遺伝子(ATP Binding Cassette Subfamily D Member 4)は、第14番染色体の長腕(14q24.3)に位置し、ゲノム上で約16キロベースの領域に広がる19のエクソンから構成されます。翻訳されるタンパク質は606個のアミノ酸残基からなる膜内在性タンパク質で、全身のさまざまな組織で幅広く発現しています。
💡 用語解説:ABCトランスポーターとは
ABC(ATP Binding Cassette)トランスポーターとは、ATPというエネルギー分子を分解して得たエネルギーで、さまざまな物質を細胞膜や細胞内膜を通じて輸送するタンパク質の大ファミリーです。ヒトのゲノムには48種類のABCトランスポーター遺伝子が存在し、構造の特徴によってAからGまでの7つのサブファミリーに分類されます。ABCD4はそのうちの「Dサブファミリー(ALDサブファミリー)」に属します。薬の排出や脂質の輸送、ビタミンの移送など、多彩な機能を持つ分子群です。
ABCD4は同じDサブファミリーに属するABCD1・ABCD2・ABCD3と約25〜27%のアミノ酸配列同一性を共有しています。構造的には「ハーフトランスポーター」と呼ばれる形式をとっており、機能するためには2つが向かい合って「二量体」を形成する必要があります。現在の研究では、ABCD4は主にホモ二量体(ABCD4が2つ向かい合った構造)として機能すると考えられています。
💡 用語解説:ハーフトランスポーター
ABCトランスポーターは一般的に、2つの「膜貫通ドメイン(TMD)」と2つの「ATP結合ドメイン(NBD)」を持つ完全な構造(フルトランスポーター)で機能します。しかしハーフトランスポーターは、TMDとNBDをそれぞれ1つしか持ちません。そのため、同じ種類または別の種類のタンパク質と「向かい合わせで手をつなぐ(二量体化)」ことで初めて、物質を運ぶ通路を形成できます。ABCD1〜4はすべてハーフトランスポーターです。
NBD(ATP結合ドメイン)には、ATPの認識に関わるWalker AモチーフとWalker Bモチーフ、そしてABCトランスポーター特有の「ABCシグネチャーモチーフ(LSGGQモチーフ)」が含まれています。これらの領域に生じる変異は、ATP加水分解能を著しく低下させ、輸送機能の完全な喪失に直結します。遺伝子はかつて「PMP69」「P70R」「PXMP1L」とも呼ばれていましたが、現在は国際的にABCD4で統一されています。
ABCD4遺伝子の基本プロファイル
| 項目 | 内容 |
|---|---|
| 遺伝子名 | ABCD4(ATP Binding Cassette Subfamily D Member 4) |
| 染色体位置 | 14q24.3(第14番染色体長腕) |
| 遺伝子構造 | 約16 kb・19エクソン |
| タンパク質 | 606アミノ酸・ハーフトランスポーター |
| サブファミリー | ABCDサブファミリー(ALDサブファミリー) |
| 細胞内局在 | リソソーム膜(および小胞体) |
| 主な機能 | ビタミンB12(コバラミン)のリソソーム→細胞質へのATP依存性輸送 |
| 関連疾患 | cblJ型メチルマロン酸血症・ホモシスチン尿症(常染色体劣性遺伝) |
| 旧称 | PMP69・P70R・PXMP1L |
| OMIM ID | ABCD4遺伝子 #603214 / cblJ型疾患 #614857 |
2. 細胞内局在のパラダイムシフト:ペルオキシソームからリソソームへ
ABCD4研究における最大の転換点は、その細胞内局在に関するパラダイムシフトです。かつて「ペルオキシソームにいるタンパク質」と信じられていたABCD4が、実際にはリソソーム膜に局在することが判明——この発見は代謝内分泌学における重大なアップデートをもたらしました。
なぜ長年「ペルオキシソームのタンパク」と誤認されていたのか
同じABCDサブファミリーに属するABCD1・ABCD2・ABCD3は、いずれもペルオキシソーム膜に局在し、極長鎖脂肪酸などをペルオキシソーム内へ取り込む働きを持つことで知られていました。ABCD4のアミノ酸配列がこれらと類似していたため、長年「69 kDaペルオキシソームABCトランスポーター(PMP69)」として分類され、ペルオキシソームに存在すると推定されていたのです。
💡 用語解説:ペルオキシソームとリソソームの違い
ペルオキシソームは細胞内の小器官で、脂肪酸の分解(β酸化)や活性酸素の無毒化を担います。リソソームは別の小器官で、細胞内に取り込まれたタンパク質・脂質・細菌などを分解・再利用する「廃棄物処理工場」としての役割を持ちます。外から取り込まれたビタミンB12は、まずリソソームに送られてキャリアタンパクから切り離されます。ABCD4はリソソームに存在することで、そこに閉じ込められたビタミンB12を細胞質へ解放する役割を担っています。
詳細な解析の結果、ABCD4にはペルオキシソームへの標的化シグナル(N末端疎水性シグナルモチーフ)が完全に欠如していることが証明されました。ペルオキシソームへの輸送に必要な「通行手形」を持っていないのです。実際の蛍光タンパク質を用いた細胞イメージング実験でも、ABCD4はペルオキシソームではなく小胞体とリソソームに局在することが確認されています。
LMBD1との複合体形成がリソソームへの「通行証」
ABCD4が小胞体で合成された後、リソソームへと正しく届けられるためには、別のタンパク質であるLMBD1との物理的な結合が不可欠です。ABCD4を単独で細胞に過剰発現させると、タンパク質は小胞体内に留まったまま、最終目的地であるリソソームへは到達できません。
💡 用語解説:LMBD1(LMBRD1遺伝子産物)
LMBD1は、LMBRD1遺伝子によってコードされるリソソーム膜タンパク質で、ABCD4の「シャペロン兼リソソーム標的化受容体」として機能します。LMBD1は小胞体でABCD4と強固な複合体を形成し、その誘導によってABCD4はエンドソーム経路を経てリソソームへと輸送されます。LMBRD1遺伝子の変異はcblF型、ABCD4遺伝子の変異はcblJ型という別々の遺伝性疾患を引き起こしますが、LMBD1-ABCD4複合体が機能しなくなる点で病態は完全に一致します。
このトラフィッキング経路はエンドソーム系を経由します。蛍光標識されたABCD4は、初期エンドソーム・後期エンドソームのマーカーと部分的に共局在することも確認されており、オートファジー経路との関連も示唆されています。臨床的に重要なのは、ABCD4またはLMBRD1のどちらかに変異があっても、リソソームへのB12輸送が止まり、同じ代謝異常を引き起こすという点です。これが、cblF型(LMBRD1変異)とcblJ型(ABCD4変異)が生化学的に区別できない理由です。
3. ビタミンB12輸送の分子機構:リソソームからの「脱出」
ABCD4の確立された主要な生理的役割は、リソソーム内腔から細胞質へのビタミンB12(コバラミン)のATP依存性輸送です。この仕組みを理解するには、まずビタミンB12が体内でどのような経路をたどるかを知る必要があります。
💡 用語解説:コバラミン(ビタミンB12)
コバラミン(ビタミンB12)はコバルトを中心に持つ巨大な水溶性ビタミンで、ヒトは自分では合成できず食事(主に動物性食品)からのみ摂取します。体内では「アデノシルコバラミン(AdoCbl)」と「メチルコバラミン(MeCbl)」という2つの活性型に変換され、それぞれミトコンドリアと細胞質の重要な代謝酵素の補酵素として機能します。この変換が起きる前に、まずリソソームから細胞質に「脱出」させる必要があり、その役割を担うのがABCD4です。
ビタミンB12の細胞内取り込みから利用までの経路
血中でビタミンB12はトランスコバラミンII(TCII)というキャリアタンパク質と結合して運ばれます。細胞はこの複合体をCD320という受容体を介した受容体介在性エンドサイトーシスによって取り込みます。取り込まれたエンドソームはリソソームと融合し、リソソーム内の強酸性環境と分解酵素(カテプシンなど)がTCIIを分解——遊離のコバラミンがリソソーム内腔に放出されます。
しかし、遊離のコバラミンは非常に大きく親水性が高いため、リソソームの脂質二重層を自力で通過することができません。ここで登場するのがABCD4-LMBD1複合体です。
💡 用語解説:ABCD4のユニークな輸送方向性(インポーター)
ABCトランスポーターの多くは「エキスポーター」として機能し、細胞内から外部へ、またはオルガネラ内へと物質を「排出」します。ABCD1〜3も同様に細胞質からペルオキシソーム内腔へ物質を取り込ませます。しかしABCD4は逆方向——リソソーム内腔から細胞質へ物質を「取り込む」インポーターとして機能します。これは哺乳類のABCトランスポーターとしては極めて異例の性質で、なぜ同じファミリー内で輸送方向が逆転しているのか、構造生物学上の大きな謎として研究が続いています。
リポソーム再構成実験による輸送能の直接証明
精製したヒトABCD4タンパク質を人工脂質二重層(リポソーム)に組み込んだ再構成実験では、ABCD4がLMBD1なしで単独でも、ATP加水分解に依存してコバラミンをリポソーム外へ輸送できることが証明されました。つまりABCD4こそが輸送の「本体」であり、LMBD1はリソソームへの正しい届け先を確保するための「案内役(シャペロン)」に相当します。
リソソームから細胞質へ解放されたコバラミンは、直ちに細胞質のシャペロンタンパク質MMACHCと複合体を形成し、還元・脱アルキル化を受けたのち、アデノシルコバラミン(AdoCbl)とメチルコバラミン(MeCbl)という2つの活性型へと変換されていきます。なお、AdoCbl合成の最終ステップではミトコンドリア内のMMAB(コバラミンアデノシルトランスフェラーゼ)が働きます。MMAB遺伝子の変異はcblB型と呼ばれる別の代謝異常症を引き起こしますが、cblJ型とは原因部位が根本的に異なります。
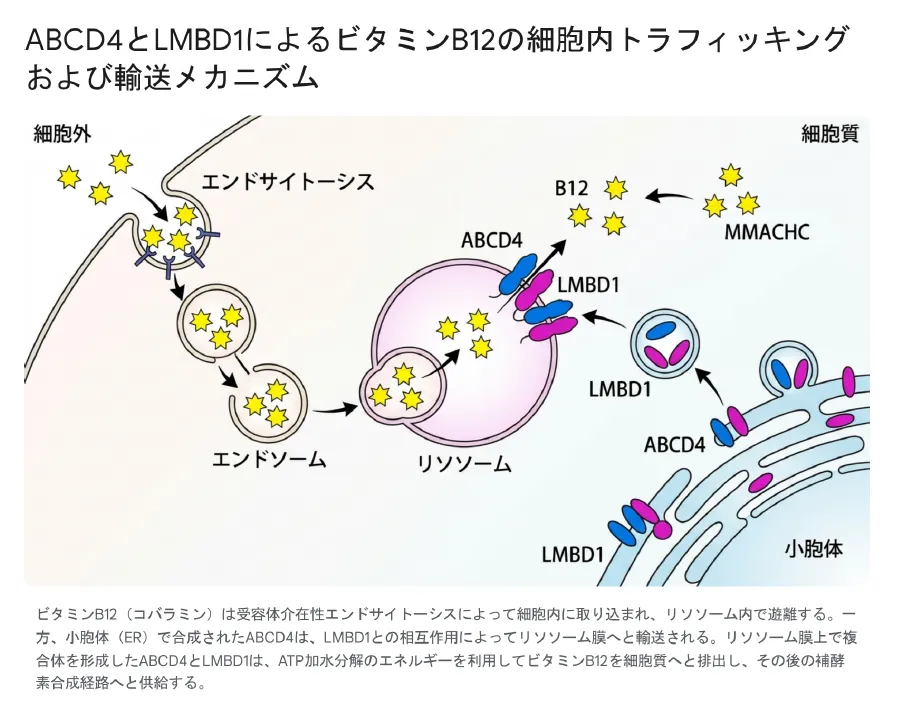
食事から吸収されたビタミンB12(コバラミン)は、リソソーム内でキャリアタンパクから遊離した後、リソソーム膜上のABCD4–LMBD1複合体によって細胞質へ輸送される。ABCD4単独でATP依存的な輸送能を持ち、LMBD1はABCD4をリソソームへ正しく届けるシャペロンとして機能する。細胞質に放出されたコバラミンはMMACHCが受け取り、アデノシルコバラミン(AdoCbl)・メチルコバラミン(MeCbl)への活性化が進む。
Cryo-EMによる構造解析:3.6Å分解能で明かされた輸送機構
2019〜2024年にかけて、Xuらの研究グループがクライオ電子顕微鏡(Cryo-EM)を用いてヒトABCD4の立体構造を3.6Åという高い分解能で解明しました(PDB ID: 6jbj)。解かれた構造は「ATP結合状態・リソソーム内腔側に開いたコンフォメーション」であり、コバラミンをリソソーム内腔から受け入れる直前のスナップショットと考えられています。
ABCトランスポーターが機能する「交互アクセスモデル」とは、ATPが結合していない状態では膜貫通ドメイン(TMD)が片側(例:細胞質側)に開いており、ATP結合・二量体化によって大規模な構造変化が起き、TMDが反対側(リソソーム内腔側)に開くというメカニズムです。ABCD4の構造データは、この動的な輸送サイクルの一瞬を捉えています。また、病的変異体(p.Asn141Lys・p.Arg432Gln・p.Tyr319Cys)が機能を失う分子的理由も、この構造データによって視覚的に証明されています。

4. cblJ型の症状と臨床スペクトラム
ABCD4の機能喪失型変異によって生じる疾患は、「cblJ型メチルマロン酸血症およびホモシスチン尿症(Methylmalonic aciduria and homocystinuria, cblJ type)」と呼ばれます。常染色体劣性遺伝の形式をとり、世界でこれまでに数十例しか報告されていない極めて稀な疾患です。
2つの代謝経路が同時に遮断されるメカニズム
ABCD4が機能しないと、リソソームに閉じ込められたコバラミンが細胞質に出てこられず、細胞全体のビタミンB12が枯渇します。その結果、2つの重要な代謝経路が同時に完全に機能不全に陥ります。
① アデノシルコバラミン(AdoCbl)経路の遮断
ミトコンドリア内の酵素メチルマロニルCoAムターゼの補酵素であるAdoCblが不足します。この酵素はバリン・イソロイシン・メチオニン・スレオニンなどのアミノ酸や奇数鎖脂肪酸の分解に必須で、機能しなくなると
→ メチルマロン酸(MMA)が体内に異常蓄積
② メチルコバラミン(MeCbl)経路の遮断
細胞質酵素メチオニンシンターゼの補酵素であるMeCblが不足します。この酵素はホモシステインをメチオニンに再合成する反応を触媒し、機能しなくなると
→ ホモシステインが蓄積・メチオニンが低下
💡 用語解説:メチルマロン酸(MMA)の毒性
メチルマロン酸(MMA)が体内に過剰に蓄積すると、高アニオンギャップ性代謝性アシドーシス(血液が酸性に傾く病態)を引き起こします。同時にミトコンドリアのエネルギー産生を直接阻害し、中枢神経系に対して強い神経毒性を発揮します。急性増悪(代謝クリーゼ)では致死的な昏睡状態に陥る危険があり、慢性的な蓄積は不可逆的な脳・腎・肝障害をもたらします。
💡 用語解説:ホモシステインの危険性
ホモシステインが血中に過剰に蓄積すると(高ホモシステイン血症)、血管内皮細胞を傷害し酸化ストレスを増大させ、若年性の重篤な血栓塞栓症のリスクを著しく高めます。また、ホモシステインの代謝産物であるメチオニンが低下すると、DNA・タンパク質のメチル化に関わるS-アデノシルメチオニン(SAM)の合成が滞り、ミエリン(神経の絶縁体)の形成不全や神経伝達物質の合成障害が生じます。
年齢層別の臨床像
🍼 胎児期・新生児期(最重症型)
- 子宮内発育遅延(IUGR)
- 非免疫性胎児水腫
- 拡張型心筋症
- 小頭症・筋緊張低下・呼吸窮迫
- 急速進行する不可逆的脳症
👶 乳幼児期(典型型)
- 成長障害・全般性発達遅滞
- 知的障害・難治性てんかん
- 代謝クリーゼ(感染時に急性増悪)
- 巨赤芽球性貧血・汎血球減少症
- 進行性網膜症・黄斑症
- 皮膚色素沈着(特徴的所見)
🧒 学童期・思春期以降(遅発型)
- 原因不明の反復性腹痛発作
- 認知機能低下・精神神経症状
- 血栓塞栓性合併症
- 亜急性連合性脊髄変性症
- しびれ・深部感覚喪失・歩行困難
最近の症例報告では、8歳で巨赤芽球性貧血を発症し反復性腹痛を主訴とする15歳の患者(p.Arg531Trp変異・ホモ接合体)が報告されており、ビタミンB12補充療法で腹痛が劇的に消失・B12レベル低下とともに再発する経過が確認されています。慢性原因不明の消化器症状とコバラミン代謝障害に直接的な因果関係があることを示す貴重な例です。
確認されている主要な病的バリアント
| タンパク質変異 | cDNA変異 | 影響を受けるドメイン・機序 | 報告背景 |
|---|---|---|---|
| p.Tyr319Cys | c.956A>G | 最後の膜貫通ドメイン(TM6)内。基質輸送経路の構造的完全性を損なう | 北米小児(フレームシフト変異との複合ヘテロ) |
| p.Glu583LeufsTer9 | c.1746insCT | 2塩基挿入によるフレームシフト。NBD構造崩壊・機能完全欠失 | 上記と同時同定 |
| p.Asn141Lys | c.423C>G | 膜貫通ドメイン(TM3)。基質認識・膜貫通ヘリックス配置に不可欠な残基 | 中国・新生児スクリーニングで同定(ホモ接合体) |
| p.Arg432Gln | c.1295G>A | Walker AモチーフのATP結合を直接阻害。能動輸送を完全停止 | ATPアーゼドメインの致命的機能不全 |
| p.Arg531Trp | c.1591C>T | エクソン17のミスセンス変異。わずかな残存活性が残る可能性 | 遅発型(15歳・反復性腹痛)から同定 |
| D143_S181del(スプライシング) | c.542+1G>T | イントロン5スプライシングドナーサイト変異。エクソン5のスキッピングを引き起こす | 欧州小児例 |
| G443_S485del(スプライシング) | c.1456G>T | エクソン14最後の塩基変換。エクソン13・14の広範なスキッピングでNBD大部分を欠失 | 欧州小児例。機能完全欠失型 |
5. 生化学的診断と相補群による鑑別診断
cblJ型の診断は、特徴的な生化学的プロファイルの確認から始まり、分子遺伝学的検査によって確定します。
診断の鍵となる3つの生化学的マーカー
MMA ↑↑
血中・尿中メチルマロン酸の著明な上昇
tHcy ↑↑
血漿総ホモシステインの著明な上昇
Met ↓/N
血漿メチオニンの低値または正常下限
重要な鑑別ポイント:ホモシステインが上昇しているにもかかわらずメチオニンが低値または正常であることは、cblJ型を含む細胞内コバラミン代謝障害の特徴です。古典的ホモシスチン尿症(シスタチオニンβシンターゼ欠損症)ではメチオニンも高値となるため、この「MMA↑ + Hcy↑ + Met↓/N」というパターンが重要な鑑別手がかりになります。
細胞内コバラミン代謝異常症の相補群比較
細胞内コバラミン代謝異常症は、「相補群(Complementation group)」と呼ばれる分類で整理されます。cblJ型は生化学的プロファイルだけでは他の相補群と区別が難しく、遺伝子検査による確定が必須です。
| 相補群 | 原因遺伝子 | 責任タンパク質の局在 | 特記事項 |
|---|---|---|---|
| cblC型 | MMACHC | 細胞質 | 最も頻度が高い(推定1/200,000〜1/60,000) |
| cblD型 | MMADHC | 細胞質・ミトコンドリア | 変異位置により複合型または単独型に分かれる |
| cblF型 | LMBRD1 | リソソーム膜 | cblJ型と生化学的・細胞レベルで表現型が完全一致。確定には遺伝子検査が必須 |
| cblJ型 ★ | ABCD4 | リソソーム膜 | 本記事の主題。世界で数十例未満の超希少疾患 |
| epi-cblC型 | PRDX1 | — | エピジェネティックな転写抑制によりcblC型と同様の症状を呈する特異なサブタイプ |
| cblX型 | HCFC1/THAP11 | 核内 | X連鎖遺伝。主に男性に発症 |
| cblB型 | MMAB | ミトコンドリア | AdoCbl経路のみの欠陥。MMA↑のみでHcy上昇なし——この点でcblJ型と明確に異なる。B12反応性MMA |
| cblC型(複合型) | MMACHC + 他 | 細胞質 | 2つの遺伝子に変異が重なる二遺伝子性(digenic)の稀なサブタイプ |
6. 遺伝子検査のアプローチ
cblJ型の確定診断には分子遺伝学的検査によるABCD4遺伝子の両アレル性病的バリアントの同定が必須です。生化学的プロファイル(MMA↑・Hcy↑・Met↓)だけではcblF型などと区別できません。
近年では新生児マススクリーニング(タンデムマス法)によるMMAの早期検出から診断につながるケースも増加しています。ただし、cblJ型は症状の重篤度が高い疾患でもあるため、生化学的異常が見つかり次第、確定診断を待たずに治療を開始するという方針が標準的です。
ミネルバクリニックで受けられる関連遺伝子検査
7. 治療・長期管理プロトコル
cblJ型の治療目標は、MMAとホモシステインの体内蓄積を抑制し、血漿メチオニン濃度を正常範囲に維持し、急性代謝不全の発作を予防することです。欠損している輸送タンパク質そのものの修復は現時点では不可能なため、治療は代謝経路のバイパスと高用量基質補充に依存します。
治療の柱
💉 ① ヒドロキソコバラミン非経口投与【最重要】
筋肉内または静脈内注射による高用量ヒドロキソコバラミン(OHCbl)投与が治療の要です。疑った時点で診断確定を待たずに直ちに開始します。大量投与により血中B12を飽和させ、一部が代替経路を通じてMMACHCに届くことを狙います。
経口投与・シアノコバラミンは無効。絶対に代用しないこと。
💊 ② ベタイン補充
ベタイン(トリメチルグリシン)はビタミンB12に依存しない別経路でホモシステインをメチオニンに変換する酵素(BHMT)の基質です。高ホモシステイン血症の改善とメチオニン枯渇の解消を目的として投与します。通常250 mg/kg/dayを1日3〜4回に分割して経口投与します。
🍃 ③ 葉酸・ホリナート投与
葉酸代謝系への負担を軽減し、メチオニン生合成を支援するために併用されます。高ホモシステイン血症を呈する患者の代謝サポートとして位置づけられます。
⚠️ 絶対的な禁忌事項:知らないと命に関わります
- ❌亜酸化窒素(笑気ガス):絶対禁忌
亜酸化窒素はコバラミンのコバルトイオンを不可逆的に酸化し、メチオニンシンターゼ活性を完全消失させます。健常者では一時的な影響ですが、cblJ型患者に投与すると急性かつ致死的な神経系崩壊と重度骨髄抑制を招きます。歯科・外科での麻酔時に必ず主治医へ伝えてください。 - ❌低タンパク質食:絶対に行ってはいけない
「単独型メチルマロン酸血症」ではMMAの前駆体アミノ酸を制限する低タンパク食が標準治療ですが、cblJ型(複合型)に低タンパク食を適用することは絶対禁忌です。メチオニン枯渇がすでにある患者から食事性メチオニンを断つと、神経症状(ミエリン化不全)を致命的に増悪させます。年齢相応の推奨摂取量(RDA)を満たすタンパク質摂取が必須です。 - ❌シアノコバラミン製剤:cblJ型には無効
一般的なビタミンB12製剤であるシアノコバラミンは細胞内での活性化プロセスが異なるため、この病型には効果を示しません。必ずヒドロキソコバラミン(OHCbl)を使用することが求められます。
8. 遺伝カウンセリング
💡 用語解説:常染色体劣性遺伝(じょうせんしょくたいれっせいいでん)
「常染色体」とは性染色体(X・Y)以外の染色体のこと。「劣性(潜性)」とは、2本の染色体の両方に変異が揃わないと症状が出ない遺伝形式です。両親がそれぞれ1つずつ変異を持っている「保因者」の場合、子どもが発症する確率は1/4(25%)、保因者になる確率は1/2(50%)、変異を持たない確率は1/4(25%)です。cblJ型はこの常染色体劣性遺伝の形式をとります。
cblJ型の確定診断後、患者・家族には以下の内容について遺伝カウンセリングが提供されます。
- ➤再発リスクの説明:両親がともに保因者であれば、次の子どもへの遺伝リスクは1/4(25%)です。両親のABCD4遺伝子検査による保因者確認が推奨されます。
- ➤同胞(きょうだい)へのリスク評価:患者のきょうだいについても保因者検査の実施を検討します。症状のない保因者は生涯を通じて影響はありませんが、将来の家族計画に備えた情報提供が重要です。
- ➤出生前診断の選択肢:次子を望む保因者カップルに対して、羊水検査・絨毛検査などによる出生前遺伝子診断が利用可能です。既知の変異が両親で確認されていれば、確実な診断が可能です。
- ➤新生児スクリーニングの活用:日本の新生児マススクリーニングでタンデムマス法によるMMA上昇が検出されれば、早期診断・早期治療開始につながります。
- ➤長期フォローアップ体制の構築:疾患の超希少性から国内外の患者レジストリや患者団体の情報が限られています。長期的な自然歴の蓄積のためにも、専門医との継続的な連携が重要です。
9. よくある誤解
誤解① 「ビタミンB12のサプリで治療できる」
経口サプリメントや普通の注射用シアノコバラミンでは、この疾患の治療はまったく効果がありません。リソソームにトラップされたB12を解放できる輸送体(ABCD4)が失われているため、大量のヒドロキソコバラミンを非経口投与して血中濃度を飽和させる必要があります。
誤解② 「低タンパク食でMMAを下げればいい」
これはcblJ型において命に関わる誤りです。低タンパク食はビタミンB12が関係しない「単独型MMA」の治療法であり、cblJ型のようにメチオニン合成が低下している患者に適用すると神経症状を致命的に悪化させます。年齢相応のタンパク質摂取が必須です。
誤解③ 「ABCD4はペルオキシソームのタンパク」
医学論文の古い記載では「ペルオキシソーム膜タンパク(PMP69)」と表記されていることがありますが、これは過去の誤解です。詳細な実験的検証により、ABCD4は実際にはリソソーム膜に局在することが証明されています。古い記述を参照して誤った理解をしないよう注意が必要です。
誤解④ 「cblJ型はcblC型と同じ疾患」
生化学的プロファイル(MMA↑・Hcy↑)が類似していても、原因遺伝子と障害部位は異なる別の疾患です。治療方針の細部にも違いがあり、また家族への遺伝リスク評価も原因遺伝子ごとに行う必要があります。遺伝子検査による正確な相補群同定が確定診断に不可欠です。
よくある質問(FAQ)
🏥 コバラミン代謝異常・遺伝子疾患に関するご相談
cblJ型をはじめとする先天性代謝疾患・希少遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
参考文献
- [1] GeneCards. ABCD4 Gene. [GeneCards]
- [2] Wiesinger C, et al. ABC Transporter Subfamily D: Distinct Differences in Behavior between ABCD1–3 and ABCD4. Int J Mol Sci. 2016;17(10):1631. [PMC5059523]
- [3] U.S. National Library of Medicine. ABCD4 gene. MedlinePlus Genetics. [MedlinePlus]
- [4] Deme JC, et al. The lysosomal protein ABCD4 can transport vitamin B12 across liposomal membranes in vitro. J Biol Chem. 2021;297(1):100869. [PMC8113721]
- [5] NCBI Gene. ABCD4 ATP binding cassette subfamily D member 4 [Homo sapiens], Gene ID: 5826. [NCBI Gene]
- [6] Xu Y, et al. Cryo-EM structure of human lysosomal cobalamin exporter ABCD4. Nat Struct Mol Biol. 2019;26(10):947-951. PDB ID: 6jbj. [PDBj 6jbj]
- [7] Watkins D, Rosenblatt DS. Disorders of Intracellular Cobalamin Metabolism. GeneReviews®. NCBI Bookshelf. [NCBI GeneReviews]
- [8] Coelho D, et al. Clinical or ATPase domain mutations in ABCD4 disrupt the interaction between the vitamin B12-trafficking proteins ABCD4 and LMBD1. J Biol Chem. 2017;292(27):11105-11114. [PMC5512089]
- [9] Orphanet. Methylmalonic acidemia with homocystinuria, type cblJ. ORPHA:26. [Orphanet]
- [10] U.S. National Library of Medicine. Methylmalonic acidemia with homocystinuria. MedlinePlus Genetics. [MedlinePlus Genetics]
- [11] Musielak M, et al. Cobalamin J Disorder in a Teenage Boy with Recurrent Abdominal Pain. Genes (Basel). 2025;16(5). [PMC12688352]
- [12] Deng X, et al. ABCD4 is associated with mammary gland development in mammals. Front Genet. 2024;15:1356091. [PMC11103957]
- [13] NCBI MedGen. Methylmalonic aciduria and homocystinuria, cblJ type (Concept Id: C3553915). [NCBI MedGen]
- [14] Wikipedia. ABCD4. [Wikipedia]






