目次

メチルマロン酸血症およびホモシスチン尿症cblF型は、LMBRD1遺伝子の変異によって細胞内リソソームにビタミンB12が閉じ込められ、代謝に使えなくなるという独自の機序を持つ超希少遺伝性疾患です。世界で数十例しか報告されていない一方、大球性貧血・汎血球減少などの血液異常が際立って顕著で、乳幼児期から神経発達・全身発育に深刻な影響をもたらします。早期診断と適切な治療介入が生命予後と発達を直接左右します。
Q. cblF型とはどのような疾患ですか?まず結論だけ知りたいです
A. LMBRD1遺伝子の病的変異により、細胞内リソソームにビタミンB12(コバラミン)が閉じ込められ、全身の細胞で使えなくなる超希少な遺伝性代謝疾患です。メチルマロン酸の蓄積(メチルマロン酸血症)とホモシステインの蓄積(ホモシスチン尿症)が同時に生じ、血液・神経・発育に深刻な影響をもたらします。世界での報告例は数十例未満で、早期診断・早期治療が予後を決定します。
- ➤疾患の定義 → Orphanet ORPHA:79284・有病率100万人に1人未満の超希少疾患
- ➤分子メカニズム → LMBD1・ABCD4複合体の機能喪失→リソソームコバラミントラッピング
- ➤主な症状 → 大球性貧血・汎血球減少・発達遅滞・筋緊張低下・一部に先天奇形
- ➤重要な鑑別 → cblJ型(ABCD4変異)と臨床・生化学的に識別不能→遺伝子検査が唯一の鑑別手段
- ➤治療 → ヒドロキソコバラミン筋肉内注射(生涯)+ベタイン+L-カルニチン
1. cblF型とは:疾患の定義と歴史的背景
メチルマロン酸血症およびホモシスチン尿症cblF型(Methylmalonic aciduria and homocystinuria, cblF type)は、第6番染色体長腕(6q13)に位置するLMBRD1遺伝子の両アレル性変異によって引き起こされる、常染色体潜性(劣性)遺伝形式をとる先天性代謝異常症です。Orphanetには「ORPHA:79284」として登録されており、有病率は100万人に1人未満の超希少疾患です。世界の医学文献に詳細な臨床報告として記録されている症例はOrphanetのデータベースでも約15例程度にとどまり、全世界の報告例は数十例未満とされています。
💡 用語解説:常染色体潜性(劣性)遺伝とは
ヒトの染色体は通常2本ずつ対になっています。「常染色体」とは性染色体(X・Y)以外の染色体のこと。「潜性(劣性)」とは2本の染色体の両方に変異が存在して初めて発症する遺伝形式です。患者の両親はそれぞれ1つの変異アレルを持つ保因者(キャリア)ですが、通常は症状が現れません。同じ保因者カップルから生まれた次の子どもの発症リスクは理論上25%です。
細胞内コバラミン代謝異常症の歴史的な分類は、患者の線維芽細胞を用いた「体細胞相補性試験(Somatic complementation analysis)」によって確立されました。複数の代謝欠陥グループ(相補性群)が同定され、cblC・cblD・cblF・cblJ・cblXなどの名称が与えられています。
💡 用語解説:相補性群(Complementation group)とは
異なる遺伝子変異を持つ2種類の患者細胞を融合したとき、お互いの欠陥が補い合って正常に戻れば「異なる相補性群(別々の遺伝子の変異)」、戻らなければ「同じ相補性群(同一遺伝子の変異)」と判定します。cblF型はこの試験によって独立したグループとして同定された病型です。現在では次世代シーケンサーによる遺伝子解析が同等の役割を担っています。
cblF型を引き起こすLMBRD1遺伝子の病的バリアントはこれまでに約9種類しか報告されていません。同定された変異の約66.7%は小規模な欠失変異であり、スプライスサイト変異やフレームシフト変異も報告されています。いずれも機能的なLMBD1タンパク質の完全な喪失(Loss-of-function)をもたらします。
2. 原因遺伝子LMBRD1と細胞内メカニズム
cblF型の病態を根本から理解するためには、正常な細胞内コバラミン代謝の複雑な経路を把握することが不可欠です。食事から摂取したビタミンB12(コバラミン)は、胃の内因子と結合して小腸(回腸末端)から吸収された後、トランスコバラミンという輸送タンパク質と結合して全身を循環します。組織の細胞はこの複合体をエンドサイトーシスで取り込み、最終的にリソソームへ運びます。
💡 用語解説:リソソームとは
細胞内に存在する「消化・分解器官」です。細胞外から取り込まれた物質を分解酵素で処理する役割を担います。コバラミンの場合、リソソームでタンパク質が分解されて遊離コバラミンが生成されます。正常ではこの遊離コバラミンが速やかにリソソーム膜を通過して細胞質へ放出されますが、cblF型ではこの排出ステップが完全に破綻しています(リソソームトラッピング)。
LMBD1タンパク質とABCD4の協調的な輸送複合体
リソソームで生成された遊離コバラミンを細胞質へ排出するには、リソソーム膜上に存在する2種類のタンパク質の精密な連携が不可欠です。
🔵 LMBD1タンパク質(LMBRD1遺伝子産物)
脂質・疎水性低分子の輸送に関わるリポカリン膜受容体と相同性を持つリソソーム膜タンパク質。ABCD4がリソソーム膜上で安定して機能するための「エスコートタンパク質/アダプタータンパク質」として働きます。cblF型ではこのタンパク質が両アレル変異によって完全に欠損します。
🟣 ABCD4タンパク質(ABCD4遺伝子産物)
ATP結合カセット(ABC)トランスポーターの一種。自身のATPase活性を利用してコバラミンをリソソームから細胞質へ能動的に輸送する実質的なトランスポーターとして機能します。ABCD4遺伝子変異ではcblJ型が発症します。
💡 用語解説:ATP結合カセット(ABC)トランスポーターとは
ATPというエネルギー物質の分解エネルギーを使って、膜を越えて物質を輸送するタンパク質の大きなファミリーです。ヒトには約50種類のABCトランスポーターが存在し、薬物耐性・胆汁酸・コレステロール輸送など多様な役割を担います。ABCサブファミリーDについての詳細解説はこちら。
LMBD1とABCD4はリソソーム膜上で物理的に強く結合してヘテロマー複合体を形成し、一つのユニットとして機能することが近年の分子細胞生物学研究によって明らかになっています。cblF型においてLMBD1が欠損すると、この複合体が崩壊してABCD4が適切にリソソームに局在・機能できなくなり、コバラミンはリソソーム内に閉じ込められたまま細胞質へ出られなくなります(リソソームトラッピング)。
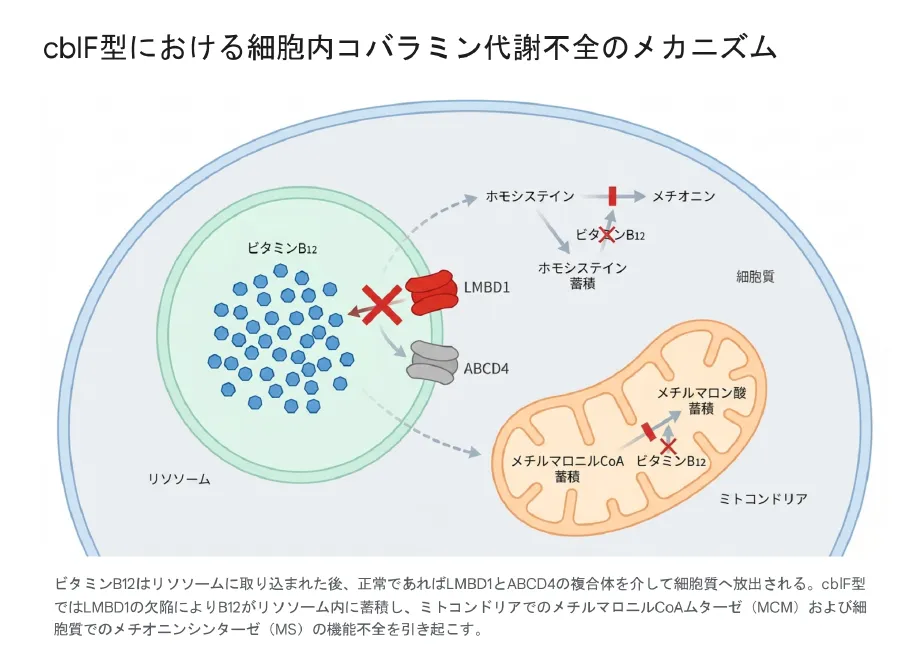
ビタミンB12はリソソームに取り込まれた後、正常であればLMBD1とABCD4の複合体を介して細胞質へ放出される。cblF型ではLMBD1の欠損によりB12がリソソーム内に蓄積し、ミトコンドリアでのMCMおよび細胞質でのメチオニンシンターゼ(MS)の機能不全を引き起こす。
二重の補酵素欠乏:メチルマロン酸とホモシステインが同時に蓄積する理由
リソソームからのコバラミン供給が断絶することで、細胞全体が重篤な機能的ビタミンB12欠乏状態に陥ります。その結果、以下の2つの重要な生化学的経路が同時に障害されます。
🔴 第一の経路:AdoCbl欠乏とMMA蓄積
コバラミンの一部はミトコンドリアでアデノシルコバラミン(AdoCbl)に変換され、メチルマロニルCoAムターゼ(MCM)の補酵素として働きます。AdoCblが枯渇するとMCMが機能を失い、メチルマロン酸(MMA)・プロピオン酸・プロピオニルカルニチンが有毒レベルで血中・尿中に蓄積します。これが重篤な代謝性アシドーシスや神経毒性を引き起こします。
🟡 第二の経路:MeCbl欠乏とHcy蓄積
コバラミンの別の一部は細胞質でメチルコバラミン(MeCbl)に変換され、メチオニンシンターゼ(MS)の補酵素として働きます。MeCblが枯渇するとMSが機能を失い、ホモシステインがメチオニンへ再変換されず血中・尿中に異常蓄積します。同時にメチオニン・SAMが枯渇し、ミエリン合成など広範な代謝に影響します。
💡 用語解説:S-アデノシルメチオニン(SAM)とは
メチオニンから合成される「生体内の主要なメチル基供与体」です。SAMはタンパク質・脂質・DNA・神経伝達物質など無数の生体分子のメチル化反応に不可欠で、特にミエリン(神経の絶縁体)の合成・維持に深く関与しています。SAMが枯渇すると神経系の発達・機能・構造が全体にわたって損なわれます。
3. 主な症状と臨床的表現型
cblF型の臨床像は個々の患者間で非常に多様であり、発症年齢は出生直後の新生児期から学童期まで幅広く報告されています。最も典型的なのは乳幼児期に発症して急速に重篤化するパターンです。胎児期にすでに影響を受けていることが多く、子宮内発育遅延(IUGR)やSGA(在胎不当過小)として出生する割合も高くなっています。
新生児期・乳幼児期の主要な症状
🩸 血液学的異常(最も顕著)
- 大球性貧血・巨赤芽球性貧血(高頻度)
- 好中球減少症・血小板減少症
- 汎血球減少症(重篤な感染症リスク増大)
- 顔面蒼白・疲労感・食欲不振
- 一部に非定型溶血性尿毒症症候群(aHUS)
🧠 神経学的症状
- 重度の筋緊張低下(フロッピーインファント)
- 嗜眠状態(極度の活動低下)
- 全般的発達遅滞(ほぼ全例)
- 脳症・てんかん発作
- 進行性の小頭症
📏 発育・全身症状
- 哺乳不良・体重増加不良
- 成長障害(Failure to thrive)
- 口内炎・舌炎・難治性皮膚発疹
- 再発性の重症感染症
🔬 先天奇形(一部の症例に合併)
- 先天性心疾患(心室中隔欠損症など)
- 三角頭蓋・口蓋裂
- 小耳症・耳介低位
- 片側性腎無発生・食道閉鎖・気管食道瘻
💡 用語解説:大球性貧血・巨赤芽球性貧血
大球性貧血(Macrocytic anemia)とは、赤血球が正常よりも著しく大きくなる貧血です。ビタミンB12や葉酸が欠乏するとDNA合成が障害され、赤血球の前駆細胞が核分裂できないまま巨大化した状態で骨髄に蓄積します(巨赤芽球性貧血 / Megaloblastic anemia)。白血球・血小板も影響を受け、汎血球減少症(Pancytopenia:赤血球・白血球・血小板すべての減少)になることが多いのがcblF型の大きな特徴です。
青年期・成人期の遅発性症状
cblF型自体の遅発性発症報告は少ないですが、コバラミン代謝異常症全体の長期病理として、以下のような重篤な中枢神経合併症のリスクが存在します。
💡 用語解説:亜急性連合性脊髄変性症
脊髄の後索(感覚伝達)と側索(運動伝達)の髄鞘(ミエリン)が変性・脱落することで生じる神経変性疾患です。主な症状は下肢のしびれ・感覚異常・運動失調・筋力低下・歩行困難です。メチオニン・SAMの枯渇によるミエリン合成障害と、異常な奇数鎖脂肪酸のミエリンへの取り込みが複合的に引き起こします。

4. 鑑別診断:コバラミン代謝異常症のスペクトラム
メチルマロン酸(MMA)とホモシステイン(Hcy)の双方が上昇する「複合型コバラミン代謝異常症」には複数の病型があります。適切な治療戦略を決定するためには病型の正確な鑑別が不可欠です。
| 病型 | 原因遺伝子 | 染色体 | 局在と機能 | 特記事項 |
|---|---|---|---|---|
| cblC型 | MMACHC | 1p34.1 | 細胞質シャペロン(コバラミンの初期処理) | 複合型で最多(約80%)。早期・遅発型あり。 |
| cblC型(二遺伝子性) | 詳細はこちら | — | 二遺伝子性(digenic)の特殊型 | 二遺伝子に同時変異を持つ希少な病型。遺伝子検査で識別。 |
| cblD型 | MMADHC | 2q23.2 | 細胞質(ミトコンドリア・細胞質への振り分け) | 変異部位によって複合型・MMA単独・Hcy単独の3表現型あり |
| cblF型 | LMBRD1 | 6q13 | リソソーム膜(ABCD4のエスコートタンパク質) | 超希少(世界数十例)。血液異常・奇形合併あり。 |
| cblJ型 | ABCD4 | 14q24.3 | リソソーム膜(ABCトランスポーター本体) | cblF型と臨床的に完全同一。遺伝子検査のみが鑑別手段。 |
| cblB型 | MMAB | 12q24.11 | ミトコンドリア(AdoCbl合成酵素) | MMA単独型。B12反応性あり。詳細はこちら。 |
| cblX型 | HCFC1 | Xq28 | 核内転写共役因子(MMACHC発現を制御) | X連鎖潜性。男児に発症。難治性てんかん・脳形成異常。 |
特筆すべきcblF型とcblJ型の関係:生化学的には識別不能
cblF型(LMBD1欠損)とcblJ型(ABCD4欠損)は原因遺伝子こそ全く異なりますが、どちらも「リソソーム内へのコバラミントラッピング」という完全に同一の細胞内病態を起こすため、血液・尿検査プロファイルおよび臨床症状が酷似し、生化学的検査のみでは絶対に区別できません。正確な病型診断にはNGSターゲットパネル解析等による遺伝子レベルでの同定(LMBRD1 vs ABCD4)が唯一の決定的な鑑別手段です。遺伝カウンセリングや将来の遺伝子治療への対応のためにも、正確な病型診断が不可欠です。
5. 診断アルゴリズムと遺伝子検査
cblF型の診断は、生化学的スクリーニングから始まり、確定的な分子遺伝学的診断へと進む段階的アプローチで行われます。早期診断が生死と長期予後を直接左右するため、疑われた時点での迅速な精査が求められます。
新生児マススクリーニング(NBS)の役割
日本でも導入されているタンデム質量分析計(MS/MS)を用いた新生児マススクリーニングは、発症前または初期段階での捕捉に重要な役割を担っています。乾燥濾紙血中のプロピオニルカルニチン(C3アシルカルニチン)濃度の上昇が検出された場合、メチルマロン酸血症の可能性として精査対象となります。JSIMDのガイドラインでもC3の異常高値は迅速な確定診断アルゴリズムへの移行を求める最重要マーカーと位置づけられています。
💡 用語解説:タンデム質量分析計(MS/MS)とは
質量分析計を2台直列につないだ装置です。乾燥血液スポット(ろ紙血)から、数十種類の代謝物を1回の測定で同時に高精度に検出できます。アシルカルニチン・アミノ酸など代謝異常のマーカーを網羅的にスクリーニングでき、cblF型を含むコバラミン代謝異常症を症状が出る前に検出することが期待されます。
必須の生化学的・血液学的検査
cblF型が疑われる際に実施すべき主要検査
- ➤血中・尿中MMA定量(GC-MS法):未治療時は数十〜数千 mmol/mol クレアチニンという異常高値を示す
- ➤血漿総ホモシステイン(tHcy)定量:著明な上昇を確認。E-HODガイドラインは原因不明の神経症状・血液異常・非定型aHUS例へのtHcy測定を強く推奨(Strong Recommendation)
- ➤血漿アミノ酸分析:ホモシステイン上昇+メチオニン著明低下(枯渇)のパターンが特徴的
- ➤血清ビタミンB12・葉酸値:後天性B12欠乏や葉酸欠乏を除外するために必須。注意:cblF型では循環血中B12値が「正常」または「高値」を示すことがあり、これが診断を遅らせる落とし穴になる
確定診断:次世代シーケンサーによる分子遺伝学的検査
現在では次世代シーケンサー(NGS)技術の進歩により、LMBRD1・MMACHC・MMADHC・ABCD4などのコバラミン代謝関連遺伝子を網羅するターゲットパネル解析、または全エクソーム解析(WES)が確定診断の第一選択となっています。両アレルにおけるLMBRD1遺伝子の病的バリアント(ホモ接合体または複合ヘテロ接合体)を同定することでcblF型と確定診断できます。
6. 治療戦略と長期管理プロトコル
cblF型に対する根本的な遺伝子治療は現時点では実用化されていませんが、早期診断から直ちに開始される代謝補正療法によって、有毒代謝産物の蓄積を抑制し、生命予後と臨床症状を劇的に改善することが可能です。E-HOD(欧州ガイドライン)とJSIMD(日本先天代謝異常学会)の指針に基づいた複合的なアプローチが標準治療として実施されます。
第一選択:ヒドロキソコバラミン(OH-Cbl)の生涯にわたる非経口高用量投与
💡 なぜ「経口」では効かないのか?
cblF型ではリソソーム受容体経路そのものがLMBRD1変異によってブロックされています。このため、薬局で一般的に購入できる経口シアノコバラミン(CN-Cbl)は、正常なリソソーム経路を使えないため無効または極めて不十分です。代替的な取り込み経路(マクロファージ系貪食・細胞膜の非特異的ピノサイトーシスなど)を通じて、細胞質内に直接高濃度のB12を届けるためには、血中濃度を急激かつ持続的に高められる筋肉内注射(IM)が不可欠です。実際に月1回のシアノコバラミン投与から週1回のOH-Cbl IMに切り替えた14歳患者で代謝異常が劇的に正常化した報告があります。
投与量のプロトコル例:急性期・乳幼児では1日1 mg(固定用量)または0.3〜0.5 mg/kg/日の頻回投与(毎日〜週数回)。代謝パラメーターが安定した後は週1回〜月1回の維持投与を生涯にわたり継続します。E-HODガイドラインは、疑いが生じた時点で確定診断を待たずに直ちにOH-Cbl投与を開始することを強く推奨しています。
必須の補助療法
🔵 無水ベタイン
BHMTを介した代替経路でホモシステインをメチオニンへ変換し、有毒なHcyを解毒します。通常250 mg/kg/日を3回分割経口投与。⚠ 過剰投与による血中メチオニン異常上昇(3000 µmol/L超)は致命的な脳浮腫のリスク。定期的なメチオニン濃度モニタリングが必須。
🟢 L-カルニチン
蓄積した有毒なプロピオニルCoA・L-メチルマロニルCoAと結合してアシルカルニチンとし、尿中排泄を促進します。二次的なカルニチン欠乏症の予防にも有効です。経口サプリメントとして継続投与します。
🟣 葉酸類・メチオニン補充
活性型L-メチル葉酸・フォリン酸・ピリドキシン(ビタミンB6)などのメチル化サイクル補助薬を追加することがあります。メチオニン枯渇が著明な場合は少量のメチオニン経口補充も検討します。
食事管理と急性期(シックデイ)マネジメント
慢性期管理として、MMA産生の上流にある分岐鎖アミノ酸(イソロイシン・バリン・スレオニン・メチオニン)の摂取を適度に制限する食事療法が行われます。ただし成長期小児への過度なタンパク制限は成長障害を招くため、代謝専門医・栄養士の指導のもとRDA(推奨食事許容量)を下回らない範囲での慎重な管理が必要です。
7. 長期予後と遺伝カウンセリング
cblF型は報告例がきわめて少ないため、数十年にわたる大規模コホート研究による予後データは存在しません。しかし14歳・18歳といった長期生存症例の追跡から、以下の傾向が明らかになっています。
✅ 治療で改善が期待できること
- 血液学的異常(大球性貧血・汎血球減少)の劇的改善
- 成長障害・体重増加不良の改善
- 血中MMA・tHcyの著明な低下・安定化
- 代謝不全による急性期死亡リスクの大幅低減
- 代謝性ストローク(基底核ラクナ梗塞)リスクの低減
⚠ 現在の治療の限界:神経学的予後
- 言語発達遅滞・運動機能不全
- 学習障害・軽〜中等度知的障害の残存(年齢・治療開始時期によらず普遍的)
- 胎生期または診断前の不可逆的中枢神経損傷が関与
- 血液脳関門の限界:脳組織の細胞内B12欠乏を完全是正できない可能性
定期的なフォローアップと多職種連携
長期にわたる安定した生活の質(QOL)維持のため、代謝専門医を中心とした多職種連携による定期的なフォローアップが不可欠です。幼児期・学童期は年に最低2回以上のクリニック受診を推奨し、以下の評価を実施します。
- ➤生化学的・血液学的評価:血漿tHcy・血中/尿中MMA・アミノ酸分析(メチオニン)・CBC(一般血液算定)
- ➤腎機能モニタリング:進行性腎機能障害(尿細管間質性腎炎)合併リスクのため、BUN・クレアチニンを定期評価
- ➤神経発達・眼科的評価:精神運動発達評価・EEG・頭部MRI、視神経・網膜病変のスクリーニング(定期眼科検診)
- ➤心血管系評価:成人期以降の血管血栓症リスクを考慮した評価を長期ケアに組み込む
遺伝カウンセリングで伝えるべき主要な内容
- ➤再発リスク:両親はそれぞれ1つの変異を持つ保因者(キャリア)。次子の発症リスクは理論上25%。
- ➤出生前診断:既知の両アレル変異が同定されている場合は羊水検査・絨毛検査による出生前遺伝子診断が可能です。
- ➤キャリアスクリーニング:保因者(キャリア)の検査については キャリアスクリーニングとは と 米国人類遺伝学会の推奨内容 もご参照ください。
8. よくある誤解
誤解①「血清B12が正常ならB12欠乏ではない」
cblF型の核心的な特徴は「血中を流れるB12は正常・高値でも、細胞内(リソソーム)に閉じ込められて使えない」という逆説的な状態です。血清B12値のみで安心してはいけません。MMA・tHcyの同時測定が必須です。
誤解②「経口ビタミンB12を飲めば治る」
一般的な後天性B12欠乏症とは全く異なります。cblF型ではリソソーム受容体経路がブロックされているため、経口シアノコバラミンは無効です。ヒドロキソコバラミンの筋肉内注射(IM)が生涯必須です。
誤解③「cblF型とcblJ型は同じ治療でよいから病型診断は不要」
現時点では治療プロトコルが類似していますが、将来の遺伝子治療・精密医療では病型(原因遺伝子)の正確な同定が必須になります。遺伝カウンセリングの再発リスク計算にも正確な遺伝子診断が不可欠です。
誤解④「ベタインは多く飲めば飲むほどよい」
ベタインの過剰投与は血中メチオニンの異常上昇を招き、致命的な脳浮腫のリスクがあります。E-HODガイドラインは厳格なメチオニン値モニタリングのもとでの用量管理を警告しています。自己判断での増量は絶対に禁止です。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 コバラミン代謝異常症・先天性代謝疾患のご相談は
cblF型をはじめとするメチルマロン酸血症・ホモシスチン尿症に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
参考文献・ガイドライン
- [1] Orphanet. Methylmalonic aciduria and homocystinuria type cblF. ORPHA:79284. [Orphanet]
- [2] MedlinePlus Genetics. Methylmalonic acidemia with homocystinuria. [MedlinePlus]
- [3] MedlinePlus Genetics. LMBRD1 gene. [MedlinePlus]
- [4] Huemer M, et al. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J Inherit Metab Dis. 2017;40(1):21-48. [PMC5203859]
- [5] Froese DS, et al. Disorders of Intracellular Cobalamin Metabolism. GeneReviews®. NCBI Bookshelf. [NCBI Bookshelf]
- [6] Rutsch F, et al. Identification of SLC46A3 as causative for the cobalamin Cbl-deficiency type cblF. Am J Hum Genet. 2009;84(4):539-547. [PubMed]
- [7] Coelho D, et al. Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism. Nat Genet. 2012;44(11):1152-1155. [PMC5059523]
- [8] Alfadhel M, et al. A New, Atypical Case of Cobalamin F Disorder Diagnosed by Whole Exome Sequencing. Mol Syndromol. 2015;6(5):254-258. [PMC4772619]
- [9] Vilotijević-Dautović G, et al. Cobalamin F disease detected by newborn screening and follow-up on a 14-year-old patient. Mol Genet Metab. 2011;105(1):116-118. [PubMed]
- [10] Sharma AP, et al. Methylmalonic acidemia (MMA) with homocystinuria cblD & cblF types – A rare disorder of vitamin B12 metabolism in the western region of India. Int J Clin Biochem Res. 2023;10(3). [IJCBR]
- [11] 日本先天代謝異常学会(JSIMD). 新生児マススクリーニング対象疾患等診療ガイドライン2019. [JSIMD PDF]
- [12] Watkins D, Rosenblatt DS. Cobalamin J Disorder in a Teenage Boy with Recurrent Abdominal Pain Attacks: A Case Report and Literature Review. PMC. 2025. [PMC12688352]
- [13] Schiff M, et al. Eighteen-Year Follow-Up of a Patient With Cobalamin F Disease (cblF): Report and Review. Am J Med Genet A. 2012. [Ovid]






