目次

📍 クイックナビゲーション
メチルマロン酸血症およびホモシスチン尿症cblC型(OMIM #277400)は、MMACHC遺伝子の変異によって細胞内ビタミンB12(コバラミン)代謝が根本から破綻する、先天性代謝異常症の中で最も頻度の高い重篤な遺伝性疾患です。ミトコンドリアと細胞質の二つの代謝経路が同時に機能不全に陥る「二重欠損」が特徴で、中枢神経・眼・腎臓・血管など全身の多臓器に不可逆的な損傷をもたらします。新生児スクリーニングによる早期発見と、ヒドロキソコバラミンを中心とした多剤併用療法が予後を大きく左右します。
Q. cblC型とはどのような疾患ですか?まず結論だけ知りたいです
A. MMACHC遺伝子(1番染色体短腕)の変異によって細胞内コバラミン代謝が障害される、常染色体潜性(劣性)遺伝の先天性代謝異常症です。アデノシルコバラミン・メチルコバラミンの両経路が同時に機能不全となり、血中メチルマロン酸・ホモシステインの蓄積とメチオニン低下が同時に起きる「二重欠損」が最大の特徴です。細胞内コバラミン代謝障害の中で最も頻度が高く、新生児スクリーニングで早期発見が可能です。
- ➤疾患の定義 → OMIM #277400、常染色体潜性遺伝、世界発生頻度は出生6〜20万人に1人
- ➤分子メカニズム → MMACHCタンパクの機能喪失によるAdoCbl・MeCbl両経路の同時破綻
- ➤主要変異と表現型 → c.271dupA(55%・早期発症型)、c.394C>T(16%・遅発型)
- ➤臨床症状 → 神経障害・巨赤芽球性貧血・黄斑変性(72%)・血管血栓・腎障害
- ➤治療の核心 → ヒドロキソコバラミン(非経口)+ベタイン+葉酸+レボカルニチン多剤併用
1. cblC型とは:疾患の定義・歴史・疫学
メチルマロン酸血症およびホモシスチン尿症cblC型(Combined methylmalonic acidemia and homocystinuria, cblC type)は、細胞内コバラミン(ビタミンB12)代謝経路における先天性異常症の中で最も頻度が高い重篤な遺伝性疾患です。OMIM番号は#277400として登録されており、1番染色体短腕(1p34.1)に位置するMMACHC遺伝子の変異によって引き起こされる常染色体潜性(劣性)遺伝疾患です。
💡 用語解説:常染色体潜性(劣性)遺伝とは
「常染色体」とは、性染色体(X・Y)以外の1〜22番染色体のことです。「潜性(劣性)」とは、同じ遺伝子の2本(両アレル)に変異が揃って初めて発症することを意味します。つまり、父親・母親それぞれから変異アレルを1本ずつ受け継いだ子ども(ホモ接合体、または複合ヘテロ接合体)のみが発症します。変異アレルを1本だけ持つ親は「保因者(キャリア)」と呼ばれ、自身は通常無症状です。両親ともに保因者である場合、子どもへの遺伝確率は各妊娠ごとに25%です。
本疾患は1969年に初めて医学文献に報告され、細胞を用いた相補性試験の解析によってcblC群(complementation group C)として分類されてきました。原因遺伝子であるMMACHC遺伝子が同定されたのは2006年のことで、それ以降、病態生理の解明が飛躍的に進みました。細胞内コバラミン代謝障害にはcblAからcblXまでの複数の病型が存在しますが(HCFC1遺伝子変異によるX連鎖遺伝のcblX型を除きすべて常染色体潜性遺伝)、cblC型は群を抜いて最も頻度が高い型です。
発生頻度については、従来は出生20万人に1人とされていましたが、新生児スクリーニングの普及により実際はこれを上回ることが判明しています。地域によって異なり、米国ニューヨーク州では出生10万人に1人、カリフォルニア州では出生6万人に1人と報告されています。この地域差は、後述する特定の遺伝子変異の創始者効果(Founder effect)や民族的背景の多様性に起因します。
💡 用語解説:創始者効果(Founder effect)とは
少数の共通祖先(創始者)を持つ集団において、特定の遺伝子変異が世代を経るごとに高頻度で蓄積・伝播する現象です。例えばcblC型では、アジア系インド人・パキスタン人・中東系集団でc.394C>T変異が特異的に高頻度に見られますが、これは歴史的に同一の祖先に由来する変異が地理的・文化的に閉じた集団内で広まったためと考えられています。
cblC型以外の複合的な細胞内コバラミン代謝障害(cblD型・cblF型・cblJ型など)は極めて稀であり、医学文献における報告例はそれぞれ20例未満にとどまっています。cblC型の特徴を理解するうえでは、これらの稀少な類縁疾患との比較も重要です。
2. MMACHC遺伝子と細胞内コバラミン代謝の分子メカニズム
cblC型の病態を根本から理解するには、MMACHC遺伝子がコードするタンパク質の役割と、それが破綻したときに引き起こされる二重の代謝障害を把握することが不可欠です。
MMACHCタンパク質の機能:コバラミンの「門番」
💡 用語解説:コバラミン(ビタミンB12)とは
コバラミンはコバルトを中心に持つ水溶性ビタミンです。食事から摂取されたコバラミン(主にシアノコバラミンやヒドロキソコバラミン)は、細胞内でMMACHCタンパク質の働きにより修飾・加工され、最終的に2種類の活性型補酵素——アデノシルコバラミン(AdoCbl)とメチルコバラミン(MeCbl)——に変換されます。これらはそれぞれ異なる代謝経路で不可欠の役割を担います。
MMACHCタンパク質は、細胞内に取り込まれたビタミンB12を活性型補酵素に変換するプロセスの中核的な出発点として機能します。具体的には、シアノコバラミンからシアン基を除去する脱シアン化(Decyanation)反応と、アルキル化コバラミンの脱アルキル化反応を触媒し、後続の代謝酵素が利用できる形態にビタミンB12を修飾します。さらに、MMACHCはMMAB遺伝子産物などの代謝酵素群とも連携しながら、修飾されたコバラミンを細胞内の適切なコンパートメントへ分配・輸送する役割も担います。
系統発生学的に、MMACHCタンパク質のC末端領域は、細菌においてコバラミンの細胞内取り込みに関与するTonBタンパク質と類似の構造を持ちます。この点から、MMACHCが単なる酵素としてだけでなく、巨大分子であるコバラミンの細胞内「トラフィッキング(輸送)」に深く関与することが示されています。コバラミンのライソソームからの輸送にはABCD4遺伝子(ATP結合カセットサブファミリーD)も関与しており、これらの遺伝子の変異はcblJ型の原因となります。
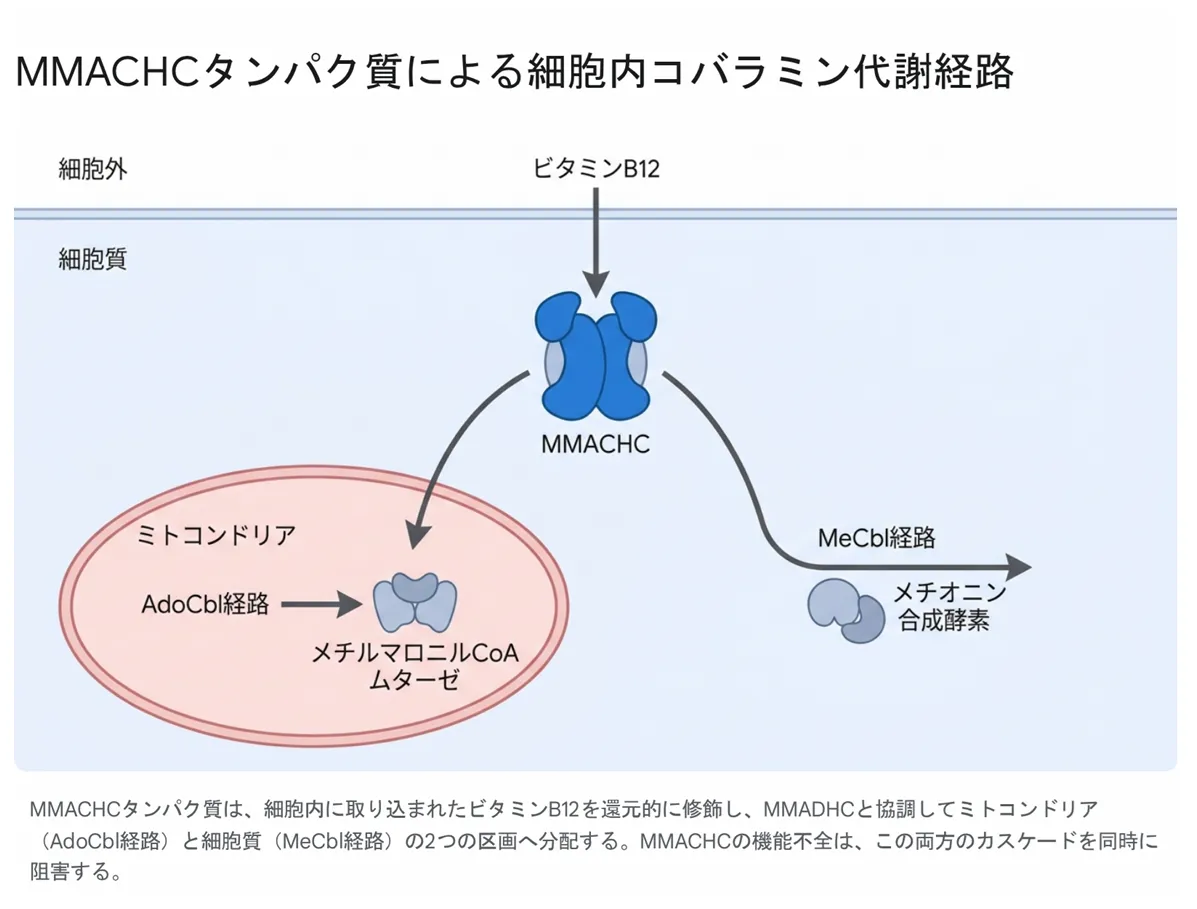
MMACHCタンパク質は、細胞内に取り込まれたビタミンB12を還元的に修飾し、MMADHCと協調してミトコンドリア(AdoCbl経路)と細胞質(MeCbl経路)の2つの区画へ分配する。MMACHCの機能不全は、この両方のカスケードを同時に阻害する。
二重欠損の生化学的カスケード
💡 用語解説:アデノシルコバラミン(AdoCbl)経路とメチルコバラミン(MeCbl)経路
AdoCbl経路(ミトコンドリア):AdoCblはメチルマロニルCoAムターゼ(MCM/MMUT酵素)の補酵素として機能します。この酵素は、バリン・イソロイシン・メチオニン・スレオニンなどの分解産物であるプロピオニルCoAをメチルマロニルCoAに経由してスクシニルCoA(TCAサイクルの中間体)へ変換します。AdoCblがないとこの変換が止まり、メチルマロン酸(MMA)が有毒なレベルで蓄積します。
MeCbl経路(細胞質):MeCblはメチオニン合成酵素の補酵素として機能し、ホモシステインにメチル基を渡してメチオニンを合成する「再メチル化反応」を担います。MeCblがないとホモシステインが蓄積(ホモシスチン尿症)し、メチオニンが低下します。
MMACHCが機能不全に陥ると、この2つの経路が同時に破綻します(二重欠損)。その結果、①血中・尿中のメチルマロン酸(MMA)・ホモシステインの異常蓄積、②メチオニン・S-アデノシルメチオニン(SAM)の産生低下、という複合的な生化学異常が同時に生じます。SAMは体内の普遍的メチル基供与体であり、その枯渇はDNAやヒストンのメチル化(エピジェネティクス制御)を含む全身のメチル化反応を低下させ、脳の白質病変や髄鞘形成不全の大きな原因となります。
近年の研究では、これらの代謝異常に加えて、細胞内酸化ストレスの増大とグルタチオン代謝の異常(レドックス・バランスの崩壊)が病態に深く関与することが判明しています。特に、正常なコバラミン自体が網膜神経節細胞(RGC)においてスーパーオキシドを消去する「内在性の神経保護物質」として機能しており、コバラミン欠乏によって局所的な酸化ストレスが暴走することが、難治性の網膜変性を引き起こす根本メカニズムと考えられています。こうした酸化ストレスへの対応を担うPRDX1(ペルオキシレドキシン1)などの抗酸化酵素系の破綻も、cblC型の重篤な眼科的合併症に寄与すると考えられています。
主要な病的バリアントと遺伝子型・表現型相関
現在までにMMACHC遺伝子では50種類以上の病的バリアントが同定されています。cblC型の大きな特徴として、遺伝子型(Genotype)と臨床表現型(Phenotype)の間に明確かつ予測可能な相関関係が存在することが挙げられます。
主要MMACHC遺伝子変異の対立遺伝子頻度と発症時期
早期発症型に関連
フレームシフト変異。ホモ接合またはナンセンス変異との複合ヘテロ接合で例外なく早期発症型を呈する。
遅発型に関連
ミスセンス変異。アジア系インド人・パキスタン人・中東系に高頻度(創始者効果)。ホモ接合では遅発型を呈することが多い。
早期発症型に関連
米国ルイジアナ州のケイジャン集団・フレンチ・カナディアンに特異的。c.271dupAと同様に重篤な早期発症型と関連。
データソース:PubMed (Methylmalonic aciduria and homocystinuria, cblC type phenotype-genotype correlations), NCBI RefSeq (MMACHC gene)
これらの遺伝子型・表現型相関データは、新生児スクリーニングで発見された未発症患者の予後予測や、保因者家族への的確な遺伝カウンセリングを提供する上で不可欠な医学的基盤となっています。なお、2つの異なる遺伝子の変異が組み合わさって発症する「二遺伝子型(Digenic)」のcblC型についても報告があります(メチルマロン酸尿症・ホモシスチン尿症cblC型(Digenic))。

3. 主な症状:早期発症型と遅発型
cblC型の臨床像は発症時期・罹患臓器・重症度において極めて多様(ヘテロジニアス)です。臨床的には発症時期に基づき「早期発症型(Early-onset)」と「遅発型(Late-onset)」に大別されます。
早期発症型(全症例の大部分を占める)
早期発症型は新生児期〜生後6ヶ月以内に急激に症状が現れます。介入の遅れは即座に生命の危機につながります。
🧠 神経系症状
- 全身性の顕著な筋緊張低下(Hypotonia)
- 難治性の痙攣発作
- 小頭症(脳発達の停滞を反映)
- 大脳萎縮・水頭症・白質病変(MRI所見)
- 精神運動発達遅滞・知的障害(後遺症)
👶 全身・消化器症状
- 重度の哺乳不良・摂食困難
- 著しい成長不良(Failure to thrive)
- 持続的な傾眠・嗜眠状態
- 胎児期からの発育不全(一部の症例)
- 代謝性アシドーシスによる急性増悪
🩸 血液系症状
- 巨赤芽球性貧血(高頻度)
- 大球性貧血・著明な蒼白(Pallor)
- DNA合成障害による赤血球分裂遅延
👁️ 眼科的合併症
- 眼振:64%、斜視:52%
- 視神経乳頭蒼白:68%
- 網膜血管狭細化:64%
- ブルズアイ黄斑変性症:72%(最重要合併症)
- 網膜外顆粒層の菲薄化(OCT所見)
💡 用語解説:巨赤芽球性貧血(きょせきがきゅうせいひんけつ)
DNA合成に必要な葉酸・コバラミン代謝が障害されると、骨髄での赤血球の細胞分裂が遅延し、異常に巨大な赤芽球(巨赤芽球)が骨髄に蓄積する状態です。血液検査では赤血球の平均容積(MCV)が増大した「大球性貧血」として現れ、著明な蒼白(顔色の悪さ)が初期の臨床的手がかりになることがあります。cblC型では、メチルコバラミン欠乏によるメチオニン合成酵素の機能不全が、葉酸代謝(メチルトラップ現象)をも障害することで巨赤芽球性貧血を引き起こします。
特に眼科的合併症は深刻です。黄斑変性は適切な薬物療法で血中代謝パラメーターが十分にコントロールされていても、全身状態とは独立して非可逆的に進行し続けるという厳しい現実があります。最終視力が法定盲レベル(20/200未満)に至る症例が55.6%を超えるという報告もあります。
遅発型(Late-onset):診断が遅れやすい特異的な症状
c.394C>T変異のホモ接合体に多い遅発型は、小児期後期〜成人期になって初めて症状が現れるため、他の神経・精神疾患として誤診されやすいことが大きな課題です。
💡 用語解説:亜急性連合性変性症(Subacute Combined Degeneration)
ビタミンB12欠乏症に典型的な脊髄病変で、脊髄の後索(感覚・位置覚)と側索(運動)の脱髄(髄鞘の破壊)が同時に進行する病態です。「連合性」とは複数の索が同時に障害されることを意味します。症状は下肢のしびれ・感覚異常・筋力低下・歩行障害・転倒として現れます。遅発型cblC型において最も特徴的な神経学的所見であり、診断のきっかけになることがあります。
遅発型の主な症状としては、①亜急性連合性変性症による感覚異常・歩行障害、②大脳白質病変・脳萎縮による認知機能障害や若年性認知症様症状、③幻覚・妄想・著しい易怒性などの急性精神症状(精神病症状)が前面に出ることがあります。さらに深刻なのが血管系合併症で、蓄積したホモシステインが血管内皮細胞を傷害し、致死的な血管血栓イベントを急発症するリスクが高い点です。
💡 用語解説:非定型溶血性尿毒症症候群(aHUS)
微小血管内に血栓が形成され、赤血球が物理的に破壊されながら腎臓の微小血管が閉塞する状態です。溶血性貧血・血小板減少・急性腎不全が三主徴で、cblC型(特に遅発型)において蓄積したホモシステインが凝固系を活性化させることで発症します。放置すれば致死的となりますが、ヒドロキソコバラミン治療への反応性が良好であるため、「原因不明のaHUS」では必ずコバラミン代謝障害を除外することが推奨されています。
4. 鑑別診断:類縁疾患との見分け方
cblC型と類似した生化学的・臨床的所見を示す疾患との鑑別は、治療方針を決定するうえで非常に重要です。
他のコバラミン代謝障害
cblA型・cblB型(cblB型詳細):MMAのみが上昇し、ホモシステイン蓄積がない点でcblC型と鑑別。
cblD・cblF・cblJ型:MMAとホモシステインが両方上昇するが、稀少で遺伝子同定による確定診断が必須。
プロピオン酸血症・MMA(mut型)
MMAが上昇するがホモシステインは正常。MMUT遺伝子変異(mut型MMA)や他の有機酸血症との鑑別には、tHcy測定と遺伝子検査が必須です。
古典的ホモシスチン尿症(CBS欠損症)
ホモシステインが高値となるがMMAは正常。CBS(シスタチオニンβ合成酵素)遺伝子の変異が原因で、水晶体脱臼・骨格異常・血栓症が特徴的。MMA正常が鑑別の決め手。
MCAD欠損症
新生児スクリーニングでC3やアシルカルニチン異常が出る別の代謝疾患(ACADM遺伝子の変異)。ホモシステイン・MMAは正常であり、アシルカルニチンプロファイルで鑑別可能。
鑑別の実際:血液・尿中のMMA・総ホモシステイン(tHcy)・メチオニンの3点セットを測定することが、cblC型を含むコバラミン代謝障害の鑑別における最も効率的なアプローチです。MMA↑かつtHcy↑かつメチオニン↓という組み合わせは、cblC型(またはその類縁病型)を強く示唆します。
5. 新生児スクリーニングと遺伝学的診断
本疾患が持つ不可逆的な進行リスクを考えると、症状が現れる前(発症前)の確定診断と日単位での迅速な治療開始が、神経学的予後を大きく左右します。拡大新生児スクリーニング(Expanded Newborn Screening: NBS)がその要となります。
一次スクリーニング:タンデム質量分析(MS/MS)
💡 用語解説:タンデム質量分析計(MS/MS)
2台の質量分析計を直列(タンデム)に連結した装置で、乾燥濾紙血(DBS: Dried Blood Spot)のごく少量のサンプルから、数十〜数百種類の代謝物を同時に超高速で測定できます。新生児のかかとから採血した血液を乾燥濾紙に染み込ませたDBSを用い、先天性代謝異常症全般のスクリーニングを効率よく行います。cblC型では、プロピオニルカルニチン(C3)の上昇が主要な一次マーカーとなります。
一次スクリーニングでは、プロピオニルカルニチン(C3)の上昇を主要マーカーとして検出します。診断特異度を高めるために、C3/C2(アセチルカルニチンとの比)、C3/C0(遊離カルニチンとの比)、C3/Met(メチオニンとの比)なども評価されます。特にcblC型では、メチルコバラミン産生低下に伴い血中メチオニン濃度が低値〜正常低値となる傾向があり、C3/Metの上昇は他のプロピオン酸血症との鑑別において高い有用性を持ちます。
セカンドティア(二次)検査:見逃しを防ぐ
一次スクリーニングには重大な限界があります。遅発型や軽症バリアントを持つ患者では、C3の上昇がカットオフ値を下回る偽陰性が生じるケースがあります。この問題を解決するために、一次検査で少しでも異常が疑われた場合に同一DBSサンプルを用いて直ちに実施する「セカンドティア検査」の導入が強く推奨されています。
🔬 セカンドティア検査で測定する項目
- ➤メチルマロン酸(MMA):カットオフ値は2 µmol/L(イタリア・ラツィオ州のNBSセンターの基準)
- ➤総ホモシステイン(tHcy):再メチル化障害の直接的マーカー
- ➤メチルクエン酸(MCA):プロピオニルCoAの蓄積を反映する補完マーカー
イタリアの大規模後方視的コホート研究では、一次検査(生後8〜20日目の再検査)でC3が完全に正常値に戻った乳児3名において、セカンドティアのMMA値のみが明確にカットオフを超えていたケース(3.1〜7.6 µmol/L)が発見されました。これにより、従来の基準では完全に見逃されていたはずの軽症・遅発型cblC患者3名が発症前診断に至りました。このエビデンスは、セカンドティアMMA/tHcy検査の実装が偽陰性を防ぐための不可欠な戦略であることを強力に裏付けています。
遺伝学的確定診断と出生前診断
💡 用語解説:次世代シーケンシング(NGS)による確定診断
NGS(Next Generation Sequencing)とは、DNAの塩基配列を網羅的かつ高速に読み取る手法です。生化学的スクリーニングで本疾患が強く疑われた場合、ゲノムDNAからMMACHC遺伝子の全コーディング領域・スプライス部位の塩基配列解析とコピー数多型(CNV)解析を行い、病的バリアントを同定して診断を確定します。確定と同時に、遺伝子型・表現型相関モデルを適用して発症時期・合併症リスクの予後予測も立てることができます。ターゲット遺伝子パネル検査も有用です。
過去に罹患児を出産した既往のある保因者家系などでは、リスク妊娠における出生前診断が標準的に提供されます。妊娠中期の羊水上清中のMMA・tHcyの生化学的測定、またはより早期に実施可能な絨毛採取(CVS)による胎児細胞DNAの直接変異解析が含まれます。既知の変異が同定されている場合、CVSでの遺伝子解析は費用対効果が高く迅速であり、出生前治療(後述)を実施する上での重要な判断基準を提供します。
6. 治療・長期管理:E-HODガイドラインに基づく多剤併用療法
cblC型の臨床マネジメントには、欧州の専門家ネットワークであるE-HOD(European Network and Registry for Homocystinurias and Methylation Defects)が策定した包括的なコンセンサスガイドライン(2017年発表、定期的に更新)が現在の国際的なゴールドスタンダードとして機能しています。
核心:ヒドロキソコバラミンをすぐに・毎日・注射で
E-HODガイドラインの最も重要な推奨事項は、コバラミン関連の再メチル化障害が少しでも疑われた段階で、確定診断を待たず一切の遅延なくヒドロキソコバラミン(OHCbl)の非経口投与を開始することです。この迅速な初期対応が生存率の劇的な改善と重篤な急性合併症の回避に直結します。
💡 用語解説:なぜヒドロキソコバラミン(OHCbl)でなければならないのか
一般的なビタミンB12製剤のシアノコバラミン(CNCbl)は、cblC型患者には一切効果がありません。シアノコバラミンを細胞内で利用するためには、MMACHCタンパク質による「脱シアン化」が必要です。しかしcblC型ではMMACHC自体が機能しないため、シアノコバラミンはまったく加工されず代謝経路に入れません。一方、ヒドロキソコバラミンは脱シアン化ステップを必要とせず代謝経路に直接入れるため、必ず筋肉内(IM)・皮下(SQ)・静脈内(IV)注射による非経口投与が要求されます。
以下は、E-HODガイドラインに基づくcblC型の標準的な薬物療法プロトコルです。
| 薬剤名(投与経路) | 推奨用量(E-HOD基準) | 投与頻度 | 主な薬理学的目的 |
|---|---|---|---|
| ヒドロキソコバラミン(IM・SQ・IV) | 0.3 mg/kg/日以上 | 1日1回(毎日) | 血中MMA・tHcy低下、メチオニン正常化、巨赤芽球性貧血改善 |
| ベタイン(経口) | 250 mg/kg/日 | 1日3回に分割 | 肝臓のBHMT経路活性化によるホモシステインの代替的再メチル化 |
| フォリン酸/葉酸(経口) | 5〜15 mg/日 | 1日2〜3回に分割 | 破綻したメチル化サイクルの維持・メチル基供給 |
| レボカルニチン(経口) | 50〜100 mg/kg/日 | 1日3回に分割 | 有毒なプロピオニル基の尿中排泄促進、二次性カルニチン欠乏の是正 |
食事療法については、歴史的に厳格な低タンパク質食が試みられてきましたが、現在のE-HODガイドラインでは過度なタンパク制限の有効性には強い疑問が呈されています。厳格な食事制限でも代謝性代償不全のリスクは排除できず、むしろ成長期の小児において必須アミノ酸欠乏や医原性の栄養不良を招く危険性が高いため、薬理学的治療による生化学的パラメーターのコントロールを主体とする方針が主流です。
胎児期からの出生前治療アプローチ
生後直後から治療を開始しても既に重篤な神経・眼科的ダメージが進行しているケースが多いことから、胎児期から介入する出生前治療が最前線のアプローチとして注目されています。過去の報告では、妊娠15週目から母親にOHCbl(週30mg筋肉内投与)と葉酸を分娩まで継続した結果、出生した新生児はアプガースコアが極めて良好(1分後9、5分後・10分後ともに10)で、生後3ヶ月時点の代謝学的フォローアップも完全に正常範囲を維持したケースが報告されています。この知見は、将来的な標準治療パラダイムになる可能性を強く示唆しています。
臓器移植(肝移植・肝腎同時移植)の意義と限界
頻繁なアシドーシス発作や代謝性代償不全を繰り返す重症患者には、臓器移植(単独肝移植:LT、または肝腎同時移植:CLKT)が選択肢となります。
メチルマロン酸血症における臓器移植の有効性比較(LT vs CLKT)
代謝性代償不全発作の根絶率
慢性腎臓病(CKD)の寛解率
移植術は代謝性代償不全の根絶において極めて有効(ほぼ100%)であるが、腎機能の長期的な寛解(CKD remission)を目指す場合、肝腎同時移植(CLKT)が単独肝移植(LT)を有意に上回る効果を示す。Data sources: ResearchGate(Safety and efficacy of liver transplantation for methylmalonic acidemia: A systematic review and meta-analysis)
しかし、臓器移植はcblC型の「完全な治癒」を意味しないという重大な限界があります。移植によって肝臓での代謝能は正常化されますが、中枢神経系や網膜細胞などが保有するMMACHC変異は修復されていないため、神経学的損傷の進行や視力低下のリスクは排除できません。このため、移植手術後もヒドロキソコバラミン等の代謝補正薬の生涯にわたる継続投与が必須です。
次世代治療:EPI-743(Vatiquinone)と遺伝子治療
酸化ストレスを直接標的とする低分子化合物EPI-743(一般名:Vatiquinone)の第2相臨床試験が実施されており、特に非可逆的な網膜変性症および神経障害への有効性が検証されています。同時に、2024〜2025年にかけてAAVベクターやレンチウイルスベクターを用いたin vivo遺伝子治療の前臨床研究が急速に進展しており、将来的な根本治療(Curative療法)としての期待が高まっています。イタリアのGenespire社とSan Raffaele Telethon遺伝子治療研究所の共同チームが開発した「免疫シールド型レンチウイルスベクター(ISLV)」を用いた前臨床データは、2025年5月の米国遺伝子細胞治療学会(ASGCT)で発表され高い注目を集めています。
7. 遺伝カウンセリングと家族支援
cblC型の確定診断後は、患者本人と家族への丁寧な遺伝カウンセリングが不可欠です。常染色体潜性遺伝であることから、保因者である両親への対応が特に重要になります。
- ➤再発リスクの説明:両親がともに保因者の場合、各妊娠で25%の確率で罹患児が生まれます。次子計画に際しては、出生前遺伝子診断の選択肢(羊水検査・絨毛検査による変異解析)について説明します。
- ➤保因者診断の検討:患者の兄弟姉妹が保因者かどうかを確認することも重要です。将来の家族計画に備えるためのキャリアスクリーニングについて説明します。
- ➤ACMG/ACOGの推奨:米国人類遺伝学会(ACMG)および米国産婦人科学会(ACOG)は、妊娠前または妊娠早期のキャリアスクリーニングを推奨しています。詳細はACMG/ACOGの推奨内容をご参照ください。
- ➤長期的な心理社会的サポート:毎日の注射スケジュール遵守・頻繁な通院・食事管理は保護者にとって大きな精神的・身体的負担となります。患者支援団体との連携や学校・社会への疾患理解促進のための教育ツール活用も重要です。
- ➤遺伝性代謝疾患の情報収集:本疾患は希少疾患であるため、国内外の患者レジストリや最新の治療情報を継続的に収集することが重要です。当クリニックでは遺伝カウンセリングを通じた長期的な情報提供を行っています。
8. よくある誤解
誤解①「市販のビタミンB12で代替できる」
市販品の多くはシアノコバラミンです。cblC型患者ではMMACHCが機能しないため、シアノコバラミンはまったく代謝できません。必ずヒドロキソコバラミンの非経口投与(注射)が必要で、経口ではなく注射であることも重要です。
誤解②「血液検査が正常になれば眼は大丈夫」
代謝パラメーターが十分にコントロールされていても、網膜変性は全身状態とは独立して進行し続けることが確認されています。定期的な眼科的精査(OCT等)は治療中も欠かせません。
誤解③「肝移植で完治する」
肝移植は代謝発作を根絶する強力な効果を持ちますが、神経・網膜細胞のMMACHC変異は修復されません。移植後もヒドロキソコバラミンの生涯投与が必要であり、「完治」ではありません。
誤解④「遅発型は軽症だから急がなくていい」
遅発型でも致死的な血管血栓(aHUS・肺動脈高血圧症)が急発症するリスクがあります。症状が出てからでは手遅れになる場合もあり、診断後は速やかな治療開始が必要です。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 コバラミン代謝異常・遺伝性代謝疾患の遺伝カウンセリング
cblC型をはじめとする遺伝性代謝疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] Carrillo-Carrasco N, Chandler RJ, Venditti CP. Combined methylmalonic acidemia and homocystinuria, cblC type. I. Clinical presentations, diagnosis and management. J Inherit Metab Dis. 2012;35(1):91-102. [PMC4219318]
- [2] GeneDx. MMACHC Gene Analysis in Methylmalonic Aciduria and Homocystinuria, cobalamin C (cblC) Type. [GeneDx Provider Portal]
- [3] MedlinePlus Genetics. Methylmalonic acidemia with homocystinuria. [MedlinePlus]
- [4] Lerner-Ellis JP, et al. Combined methylmalonic aciduria and homocystinuria (cblC): phenotype-genotype correlations and ethnic-specific observations. Mol Genet Metab. 2006;88(4):315-321. [PubMed]
- [5] NCBI GeneReviews. Disorders of Intracellular Cobalamin Metabolism. [NBK1328]
- [6] Martinelli D, et al. Combined methylmalonic acidemia and homocystinuria, cblC type. II. Complications, pathophysiology, and outcomes. J Inherit Metab Dis. 2011;34(3):509-514. [PMC3529128]
- [7] Froese DS, et al. Versatile enzymology and heterogeneous phenotypes in cobalamin complementation type C disease. JIMD Rep. 2022;65(4):261-270. [PMC9464900]
- [8] Tsang MH, et al. Ophthalmic Manifestations and Long-Term Visual Outcomes in Patients with Cobalamin C Deficiency. Ophthalmology. 2016;123(12):2484-2495. [PMC5065014]
- [9] Huemer M, et al. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J Inherit Metab Dis. 2017;40(1):21-48. [PMC5203859]
- [10] Martinelli D, et al. Efficacy of early treatment in patients with cobalamin C disease identified by newborn screening: a 16-year experience. J Inherit Metab Dis. 2018;41(5):893-904. [PMC6082364]
- [11] Spagnoli C, et al. Successful intrauterine treatment of a patient with cobalamin C defect. Mol Genet Metab Rep. 2016;6:57-59. [PMC4789385]
- [12] Profilo MA, et al. Milder Form of Cobalamin C Disease May Be Missed by Newborn Screening. Metabolites. 2021;11(3):77. [MDPI]
- [13] Sharma AP, et al. Safety and efficacy of liver transplantation for methylmalonic acidemia: A systematic review and meta-analysis. Liver Transpl. 2021;27(4):543-554. [PubMed]
- [14] Animal models of methylmalonic acidemia: insights and challenges. PMC. 2025. [PMC12619279]
- [15] EPI-743 in Cobalamin C Defect: Effects on Visual and Neurological Impairment. ClinicalTrials.gov NCT01793090. [ClinicalTrials.gov]
- [16] Cobalamin-Associated Superoxide Scavenging in Neuronal Cells Is a Potential Mechanism for Vitamin B12–Deprivation Optic Neuropathy. Am J Pathol. 2017. [PMC5745528]






