目次

MMACHC遺伝子は、ビタミンB12(コバラミン)の細胞内代謝における「最初のゲートキーパー」として機能する極めて重要な遺伝子です。両アレルの変異によりcblC型メチルマロン酸血症・ホモシスチン尿症(OMIM #277400)が発症し、これは細胞内コバラミン代謝異常症のなかで最も頻度が高い疾患として知られています。乳児期の致死的代謝危機から、非可逆的な網膜黄斑変性、成人期に初めて顕在化する複雑な神経・精神症状まで、極めて多彩なスペクトラムを示します。本稿では分子機構から最新の治療戦略まで、臨床遺伝専門医が包括的に解説します。
Q. MMACHC遺伝子とcblC病とはどのような疾患ですか?まず結論だけ知りたいです
A. MMACHC遺伝子は、細胞内に取り込まれたビタミンB12(コバラミン)を処理し、2種類の活性型補酵素(MeCbl/AdoCbl)への変換を開始する「代謝の交差点」です。両アレルの機能喪失により、有毒なメチルマロン酸とホモシスチンが蓄積し、メチオニンが枯渇するという「二重の生化学的欠陥」が生じ、これがcblC病の全身症状の根本原因となります。
- ➤疾患の定義 → OMIM #277400、常染色体潜性遺伝、有病率約10万人に1人(NY州)
- ➤分子メカニズム → MMACHC-MMADHC複合体によるコバラミントラフィッキング
- ➤変異スペクトラム → 欧米 c.271dupA/中国 c.609G>A という地理的差異
- ➤臨床像 → 早期発症型の代謝危機と遅発型の6サブタイプ
- ➤最新治療 → OHCbl高用量療法・Vatiquinone・AAV遺伝子治療の最前線
1. MMACHC遺伝子とcblC病:疾患の定義と歴史的背景
MMACHC遺伝子(metabolism of cobalamin associated C)は、第1番染色体短腕(1p34.1)に位置する遺伝子で、細胞内に取り込まれたビタミンB12を機能的な補酵素へと変換するプロセスの「出発点」を担います。両アレルに病的バリアントが生じると、cblC型メチルマロン酸血症およびホモシスチン尿症(cblC病)が発症します。
💡 用語解説:常染色体潜性遺伝(じょうせんしょくたいせんせいいでん)
「潜性(劣性)」とは、両親から受け継いだ2本の染色体のうち、両方に変異がある場合にのみ症状が現れる遺伝形式のことです。両親は片方のアレルにのみ変異を持つ「保因者(キャリア)」で、通常は無症状。cblC病は常染色体潜性遺伝のため、両親が共に保因者である場合、子どもが発症する確率は25%(1/4)となります。保因者同士の結婚である可能性を知ることが、遺伝カウンセリング上きわめて重要です。
cblC病はOMIM #277400およびOrphanet ORPHA:79282として国際的に登録されており、細胞内コバラミン代謝異常症のなかで最も頻度の高い疾患です。米国ニューヨーク州の推計では約10万人に1人、カリフォルニア州のヒスパニック系人口では3万7,000人に1人の割合で発生しており、新生児スクリーニング(NBS)が普及した現在では、かつて想定されていたよりも高い頻度で発見されていることが判明しています。
歴史的には、1969年にMudd、Levy、Abelesらによって「ビタミン代謝の異常によって引き起こされる未知の疾患」として初めて記述されて以来、半世紀以上にわたり病態解明が進められてきました。原因遺伝子MMACHCの同定は2006年に達成され、その後の構造生物学的研究・エピジェネティック研究の進展により、疾患の分子基盤が段階的に明らかになっています。
2. MMACHCの分子機能とコバラミン代謝ネットワーク
MMACHCタンパク質は単なる結合タンパク質ではなく、極めて特異的な酵素活性を有する還元酵素です。細胞内に取り込まれた様々な形態のビタミンB12からリガンド(シアン基やアルキル基)を除去し、細胞内で相互変換可能な中間体へとプロセシングします。この処理を経なければ、後続の補酵素合成経路は一切機能しません。
💡 用語解説:ビタミンB12(コバラミン)と2つの補酵素
ビタミンB12は、細胞のエネルギー産生・脂質およびアミノ酸代謝・DNA合成・神経髄鞘の形成に不可欠な補酵素の前駆体です。細胞内では2つの活性型に変換されます。
① メチルコバラミン(MeCbl):細胞質でメチオニンシンターゼの補因子として働き、ホモシステインをメチオニンへ変換する。
② アデノシルコバラミン(AdoCbl):ミトコンドリア内でメチルマロニルCoAムターゼの補因子として、脂肪酸・分岐鎖アミノ酸・コレステロールの分解に関与する。
MMACHCの機能不全は、この両方の経路を同時に遮断する点が特徴です。
MMACHCの酵素活性:脱シアン化と脱アルキル化
生化学的解析の進展により、MMACHCタンパク質は以下の2つの還元酵素活性を併せ持つことが明らかになっています。
② アルキルコバラミン還元酵素:メチルコバラミンやアデノシルコバラミンなどのアルキル基を脱離させる。
この処理により、生体に取り込まれた多様な形態のビタミンB12がリガンド除去を経て、再利用可能な中間体(コア構造)へとプロセシングされる。
MMADHCタンパク質との複合体形成
MMACHC単独では機能は完結しません。処理された中間体を適切な細胞内部位へ配送するため、MMADHCタンパク質(cblD型の原因遺伝子産物)との直接的な相互作用が不可欠です。X線小角散乱(SAXS)および結晶構造解析により、コバラミンが存在することで両者は強固な1対1のヘテロ二量体を形成することが判明しています。
近年の構造生物学的研究は、この複合体が主にCo-S(コバルト-硫黄)配位結合を含む分子の「クラスプ(留め金)」構造によって物理的に維持されていること、複合体界面においてMMACHC上のArg-112側鎖とMMADHC上のGlu-194側鎖が約4オングストロームの至近距離で塩橋を形成している可能性が高いことを明らかにしています。この複合体は、処理されたビタミンB12を細胞質(MeCbl生成用)およびミトコンドリア(AdoCbl生成用)へ振り分ける「トラフィッキング・シャペロン」として働きます。
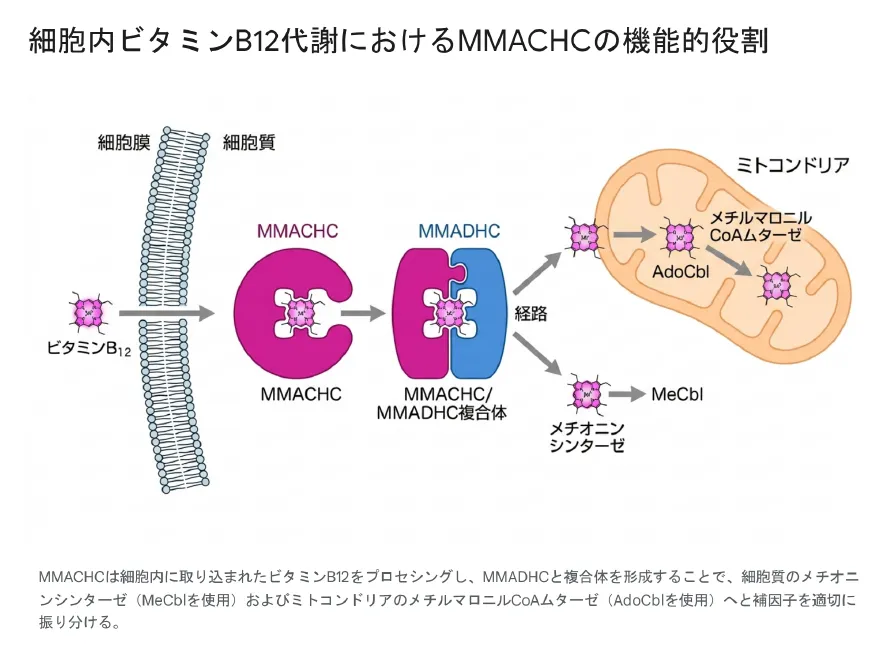
MMACHCは細胞内に取り込まれたビタミンB12をプロセシングし、MMADHCと複合体を形成することで、細胞質のメチオニンシンターゼ(MeCblを使用)およびミトコンドリアのメチルマロニルCoAムターゼ(AdoCblを使用)へと補因子を適切に振り分ける。
「二重の生化学的欠陥」という病態の本質
MMACHCが機能不全に陥ると、MeCblとAdoCbl両方の経路への補酵素供給が同時に遮断されます。その結果、体内には有毒なメチルマロン酸とホモシスチンが蓄積する一方、タンパク質合成に不可欠なメチオニンが枯渇するという「二重の生化学的欠陥」が生じ、これが多岐にわたる重篤な全身症状の根本原因となります。
変異スペクトラムと地理的特異性
cblC病を引き起こすMMACHC遺伝子の変異は、これまでに100種類以上が同定されており、人種や地域集団によって変異のスペクトラムが大きく異なります。さらに、特定の変異タイプは発症時期および臨床的重症度と強い相関を持つことが確認されています。
| 病的バリアント | 主な発症時期 | mRNA発現 | 地理的特異性 |
|---|---|---|---|
| c.271dupA | 早期発症・重症 | 著しい低発現 | 欧米集団で最多(40〜55%) |
| c.331C>T | 早期発症・重症 | 低発現 | 欧米集団で約9% |
| c.394C>T | 遅発発症 | 比較的高発現 | 遅発型に特異的(約16%) |
| c.609G>A | 早期・遅発混合 | 多様 | 中国集団で最多(約55%) |
| c.482G>A | 遅発発症 | 比較的高発現 | 欧州および中国北部(約34%) |
重症早期発症型と関連するc.271dupAホモ接合体では、対立遺伝子発現解析においてMMACHC転写産物レベルが著しく低いことが確認されています。一方、遅発型に特徴的なc.394C>TではmRNAが比較的高レベルで維持されており、この発現量の差がタンパク質の残存機能に寄与し、遅発型患者が人生後半まで致死的代謝危機を免れる生物学的根拠の一つと考えられています。
💡 用語解説:Minigeneシステムによる病原性評価
次世代シーケンサー(NGS)の普及によって多数のVUS(意義不明バリアント)が報告されるようになりました。Minigeneシステムは、患者から同定されたスプライシング変異などを対象に、プラスミドを用いて細胞モデル内で人工的に転写を行わせ、mRNAの異常スプライシングパターンを直接検証する実験手法です。2024〜2025年に有用性が実証され、特定の変異がどのようにエキソンスキッピングやイントロン保持を引き起こすかをインビトロで明確に解明することを可能にしました。
epi-cblC:PRDX1変異によるエピジェネティック沈黙
cblC病の分子遺伝学における革新的発見の一つが、MMACHC遺伝子そのものには病的バリアントが存在しないにもかかわらず同じ臨床症状を呈する「epi-cblC」という病態です。この疾患はMMACHCプロモーター領域の異常な高メチル化により、遺伝子が転写レベルで完全に沈黙(サイレンシング)することで発症します。
驚くべきことに、このエピジェネティック沈黙の原因は、第1番染色体上でMMACHCに隣接するPRDX1遺伝子の変異にあります。PRDX1のスプライシングアクセプター部位変異(c.515-1G>T または c.515-2A>T)が生じると、最終エクソンのスキッピングとポリ(A)付加シグナルの喪失が起こり、異常なアンチセンス転写リードスルーがMMACHCプロモーター領域のCpGアイランドにシス-高メチル化を誘導します。
イタリア・トスカーナ州の報告では、2001年以降に新生児スクリーニングで診断されたcblC症例の約13%がepi-cblCであったとされ、決して稀な現象ではありません。このエピ変異は生殖細胞系列で維持され、世代を超えて遺伝する可能性が報告されており、稀少疾患における二次的エピジェネティック制御の破綻を示す重要なパラダイムとなっています。

3. 主な症状と表現型スペクトラム
cblC病の臨床症状は発症時期によって「早期発症型(生後12ヶ月未満)」と「遅発型(1歳以降)」に大別されます。症状のスペクトラムは代謝系のみならず、中枢神経・末梢神経・眼科・血液・腎臓など全身のあらゆる臓器に及びます。
早期発症型:乳児期の代謝危機と非可逆的ダメージ
早期発症型は通常、生後数日から12ヶ月未満に最初の症状が現れます。初期症状は特異性に乏しく、哺乳不良・著しい体重増加不良・頻回の嘔吐・原因不明の発熱・傾眠傾向などが認められます。これらは代謝異常による有毒物質蓄積(アシデミア)に起因する急性代謝危機の現れです。
🧠 中枢神経系
- 重度の筋緊張低下(フロッピーインファント)
- 難治性のてんかん発作
- 小頭症
- 著しい発達遅滞・知的障害
🩸 血液・血管系
- 巨赤芽球性貧血
- 血小板減少・好中球減少
- 血管内皮ダメージによる微小血管障害
- 血栓リスクの飛躍的上昇
👁️ 眼科合併症
- ブルズアイ黄斑症(特徴的)
- 網膜の菲薄化・色素変性
- 視神経萎縮・斜視
- 感覚性眼振・視覚的無関心
💪 全身症状
- 著しい成長障害
- 代謝性アシドーシス
- 高アンモニア血症
- 腎血栓性微小血管症(TMA)
💡 用語解説:ブルズアイ黄斑症(雄牛の目状黄斑症)
網膜中心部の黄斑周辺が低色素となり、その周囲を高色素の輪が取り囲むことで、まるで「雄牛の目」のように見える特徴的な色素変性です。光干渉断層計(OCT)で網膜の菲薄化を伴います。cblC病における眼科合併症のなかで最も特徴的な所見の一つで、網膜電位図(ERG)を用いれば眼底検査で視覚的異常が確認される前の無症候期(例:生後数ヶ月時点)から光受容体細胞の機能低下を感度良く検出できます。
遅発型cblC病:6つの臨床サブタイプ
遅発型cblC病は1歳以降、多くは学童期から青年期、時には成人期(60代での発症例も報告)に初めて症状が顕在化します。急性代謝不全よりも緩徐に進行する神経変性疾患や精神疾患に酷似したプレゼンテーションを示すため、臨床診断が極めて困難です。
中国北部で行われた156人の遅発型cblC患者を対象とした多施設共同研究では、初発症状の優位性に基づき以下の6つの臨床サブタイプに分類されています。
遅発型cblC病の初発症状に基づく臨床サブタイプの分布
156名の遅発型cblC患者の多施設共同研究データに基づく。脳症(精神障害や認知機能低下)を主徴とするタイプが最多。
最大グループである脳症優位型(cblC-E)は、精神・行動異常、てんかん、知的・認知機能低下を主徴とし、血中総ホモシスチン(tHcy)レベルの著しい上昇と統計的に強い相関を示します。これはホモシスチン毒性が中枢神経系に直接的ダメージを与えていることを示唆しています。
遅発型cblCは精神科・神経内科・腎臓内科など複数の診療科にまたがる症状を呈するため、詳細な血中ホモシスチンおよびメチルマロン酸レベルの評価なしでは、単なる栄養性貧血・脳性麻痺・原因不明の自閉症スペクトラム障害などとして長期にわたり誤診されるリスクが高いことに注意が必要です。
網膜変性の分子基盤:酸化ストレスの関与
cblC病における最も特徴的かつ非可逆的な合併症の一つが網膜黄斑変性です。この進行メカニズムには、メチルマロン酸・ホモシスチンそのものの直接的毒性に加えて、酸化ストレスとミトコンドリアの機能不全が中心的役割を果たしています。
網膜色素上皮細胞(RPE)と視細胞は生体内で最も酸素消費量が高い細胞群であり、多価不飽和脂肪酸を豊富に含むため、光照射に伴う酸化ストレス(活性酸素種:ROS)に極めて脆弱です。MMACHCの機能不全はミトコンドリアAdoCbl産生障害を通じてエネルギー代謝を阻害し、ROS漏出を助長します。蓄積したROSは脂質過酸化・DNA損傷を引き起こし、鉄依存性細胞死(フェロトーシス)を誘導して網膜組織を不可逆的に破壊します。
4. 鑑別診断:他のコバラミン代謝異常症との違い
cblC病は「メチルマロン酸血症+ホモシスチン尿症」という生化学所見を呈する疾患群のなかで最も頻度が高いものですが、同じ生化学パターンを示す複数の疾患が存在するため、原因遺伝子レベルでの鑑別が必須です。
| 疾患型 | 原因遺伝子 | 代謝異常 | 特徴 |
|---|---|---|---|
| cblC型 | MMACHC | MMA+HCU | 最多。両経路障害。本疾患 |
| cblD型 | MMADHC | MMA・HCU・両者 | 3つの生化学サブタイプに分類 |
| cblF型 | LMBRD1 | MMA+HCU | リソソームからのB12放出障害 |
| cblJ型 | ABCD4 | MMA+HCU | cblFに類似。ATP結合カセット |
| cblX型 | HCFC1 | MMA+HCU | X連鎖。MMACHC転写制御異常 |
| cblB型 | MMAB | MMAのみ | AdoCbl合成障害。B12反応性 |
| epi-cblC | PRDX1(エピ変異) | MMA+HCU | MMACHC配列は正常。メチル化沈黙 |
| cblC digenic | MMACHC + PRDX1 | MMA+HCU | 2遺伝子モデル |
💡 用語解説:ABCDトランスポーターファミリー
ABCD4はATP結合カセット(ABC)トランスポータースーパーファミリーのサブファミリーDに属するタンパク質で、cblJ型の原因です。詳細はATP結合カセットサブファミリーDの用語解説をご参照ください。cblFのLMBRD1とともに、ビタミンB12のリソソームからの放出に関与します。
鑑別上重要なその他の代謝性疾患
新生児期の急性代謝不全を呈する疾患として、中鎖アシルCoA脱水素酵素欠損症(ACADM遺伝子)、孤立性メチルマロン酸血症(MUT欠損)、古典型ホモシスチン尿症(CBS欠損)なども鑑別に挙がります。cblC病はこれらと異なり「メチルマロン酸+ホモシスチンの同時蓄積」を特徴とするため、アシルカルニチン分析と有機酸分析の組み合わせが重要な方向付けとなります。
5. 診断基準と遺伝子検査の進め方
cblC病は重篤な非可逆的ダメージを伴い、発症後の治療介入では神経系や網膜の障害を回復させることが困難であるため、発症前または症状が極めて軽微な段階での早期発見が患者の生命とQOLを決定づけます。
新生児スクリーニング(NBS)とセカンドティア検査
世界中の多くの国や地域で、タンデム質量分析(MS/MS)によるNBSが導入されています。cblC病の場合、一次スクリーニングマーカーとして血中プロピオニルカルニチン(C3)の上昇、およびC3とメチオニン(Met)・カルニチン・アセチルカルニチンとの比率異常が用いられています。
しかし、イタリア・ラツィオ州NBSセンターの詳細解析が示すように、従来の一次マーカーへの依存は特に軽症型cblC病において重大な偽陰性リスクを抱えています。初回検査でC3異常を指摘された乳児が、2回目の乾燥血液濾紙(DBS)検査でC3値が正常範囲に低下してしまう症例が複数存在します。
💡 用語解説:セカンドティア検査(二次検査)
一次スクリーニングマーカーの異常を確認するために行う追加検査のこと。cblC病においては、C3値が正常範囲に回復していても血中メチルマロン酸(MMA)の直接測定を実施することで、カットオフ値(通常2 µmol/L)をわずかに超えるだけの軽微な生化学的異常を確実に捉えられます。MMAはcblC病における下流の主要な毒性代謝産物であるため、一次マーカーよりも疾患状態を直接的に反映します。
分子遺伝学的検査:NGSパネルからメチル化解析まで
生化学的診断後は、MMACHC遺伝子を中心とする分子遺伝学的確定診断が不可欠です。段階的に以下のアプローチが推奨されます。
💡 cblC病・関連疾患診断の検査フロー
- ➤第1段階:MMACHC遺伝子の全コード領域シーケンス(サンガー法またはNGS)
- ➤第2段階:MMA・HCU NGSパネルによる関連遺伝子網羅解析(cblD/F/J/X/B型の除外)
- ➤第3段階:MMACHC陰性の場合、MMACHCプロモーター領域のメチル化解析(epi-cblCの同定)とPRDX1遺伝子解析
- ➤第4段階:スプライシングバリアント疑いの場合、Minigeneアッセイによる機能的検証
- ➤補助検査:全エクソーム解析または核・ミトゲノムNGS検査による鑑別
6. 治療戦略:標準療法から次世代アプローチまで
cblC病の治療は、機能不全に陥った経路を迂回して欠損した補酵素を外部から補充する対症療法が主体です。しかし、特定の臓器合併症(特に眼科的合併症)の進行を完全に食い止めることは依然として困難であり、酸化ストレス制御や遺伝子レベルでの根本的アプローチの開発が急務となっています。
標準治療の柱:ヒドロキソコバラミン(OHCbl)高用量療法
標準治療の根幹をなすのが、ヒドロキソコバラミン(OHCbl)の筋肉内注射または静脈内投与です。OHCblはシアノコバラミン(市販ビタミン剤に多く含まれるが、MMACHC機能不全下ではシアン基の脱離ができないため利用困難)とは異なり、体内で利用可能な補酵素の形へと迂回して変換されやすい性質を持ちます。
💡 用語解説:なぜ市販B12サプリは効かないのか
一般的なビタミンB12サプリメントに含まれるシアノコバラミンは、活性型に変換されるためにMMACHCによる「脱シアン化」が必要です。cblC病患者はこの酵素機能そのものが失われているため、シアノコバラミンを投与しても代謝できません。一方ヒドロキソコバラミン(OHCbl)は、MMACHCを介さない代替経路を経て利用可能な中間体へ変換されやすく、cblC病の第一選択治療薬となります。
投与タイミングと投与量が予後を決定づけます。近年のエビデンスは、従来の低用量(0.3 mg/kg/日以下)よりも高用量(0.4〜2.7 mg/kg/日、一部研究では最大6.5 mg/kg/日)での強力な介入が、眼科的予後の改善およびブルズアイ黄斑症の進行遅延に有意に寄与する可能性を示唆しています。
マルチモダリティ・ケア
💊 ベタイン補充
有毒なホモシスチンをメチオニンへ変換する代替経路を促進します。神経系保護とメチオニン補充の双方に寄与。
💊 L-カルニチン補充
メチルマロン酸蓄積に伴う二次性カルニチン欠乏を補い、有機酸排泄を促進します。
🍽️ タンパク質制限食
前駆アミノ酸の負荷を軽減。ただし過度な制限は成長障害を招くため、専門管理栄養士の介入が必須。
次世代治療①:Vatiquinone(EPI-743)による酸化ストレス制御
cblC病の病態進行、特に網膜変性においてミトコンドリアのレドックス・ホメオスタシスの崩壊と過剰なROS発生が中心的役割を果たしていることから、酸化ストレスメカニズムを直接標的とする薬物療法が開発されています。その最有力候補がVatiquinone(別名EPI-743:α-トコトリエノールキノン)です。
Vatiquinoneは、脂質過酸化経路の鍵酵素である15-リポキシゲナーゼ(15-LO)のファーストインクラス選択的阻害剤で、異常な脂質過酸化やROS蓄積に起因するフェロトーシス(鉄依存性細胞死)を強力に防止します。cblC病患者特有の網膜変性および神経機能障害の進行抑制を目的として、第2相二重盲検プラセボ対照臨床試験(NCT01793090)が実施されています。
特筆すべきは、Vatiquinoneがミトコンドリア機能障害を伴う別の希少神経変性疾患であるフリードライヒ運動失調症(FA)の長期試験(MOVE-FA試験等)で、プラセボと比較して疾患進行の指標(mFARSスコア)が約50%の進行遅延を示したことです。このデータに基づき、2024年12月にFDAへ新薬承認申請(NDA)が提出され、2025年2月に優先審査指定を受けました。同様のメカニズムが関与するcblC病への応用が期待されます。
次世代治療②:AAVベースの遺伝子補充療法
疾患の根本原因である遺伝子欠損そのものを修復あるいは置換する「細胞・遺伝子治療(CGT)」分野が目覚ましく進展しています。MMACHCの下流で働くメチルマロニルCoAムターゼ(MUT)欠損に対するmRNA治療(mRNA-3704、NCT03810690)が既に臨床試験段階にあり、MMACHC自体を標的とした研究も前臨床段階で加速しています。
2023年には、CRISPR/Cas9技術を用いて患者で同定されたMMACHC変異(c.80A>G, p.Q27R)をホモ接合で持つマウスモデルの構築に成功。肝臓や神経系に高親和性を持つAAV8やAAV9ベクターを用いた生体内遺伝子補充療法の研究が進み、動物モデルでは血中メチルマロン酸濃度の劇的低下と生存率改善が確認されつつあります。2025〜2026年にかけては、AAVベクターによる希少代謝疾患・神経筋疾患へのFDA規制環境が整備されつつあり、「1回限りの治癒的アプローチ(One-and-done therapy)」への期待が極めて高まっています。
7. 遺伝カウンセリングと家族計画
cblC病は常染色体潜性遺伝のため、両親は通常無症状の保因者(キャリア)です。確定診断後の家族への丁寧な遺伝カウンセリングには以下の重要事項が含まれます。
- ➤再発リスクの説明:両親が共に保因者の場合、次子の発症確率は25%(1/4)、保因者となる確率は50%(2/4)です。
- ➤epi-cblCの遺伝形式:PRDX1関連epi-cblCではエピ変異が生殖細胞系列で維持され、一部の報告では3世代にわたる遺伝が確認されているため、従来の潜性遺伝モデルとは異なる遺伝カウンセリングが必要です。
- ➤保因者スクリーニング:血縁者や次子希望時には保因者検査を検討します。米国人類遺伝学会の推奨内容や一般的なキャリアスクリーニングのフレームワークが参考になります。
- ➤出生前診断の選択肢:両親の病的バリアントが同定されていれば、絨毛検査・羊水検査による出生前遺伝子診断が可能です。
- ➤患者家族の体験共有:他の稀少遺伝性疾患に関する体験は、家族計画の参考になります(例:ALD保因者検査 姉妹の体験談、ALDと家族計画)。
8. よくある誤解
誤解①「市販のB12サプリで補えば大丈夫」
市販ビタミンB12サプリに含まれるシアノコバラミンは利用できません。cblC病の治療にはMMACHCを介さないヒドロキソコバラミン(OHCbl)の注射投与が第一選択となります。
誤解②「生化学的コントロールができれば安心」
血中メチルマロン酸・ホモシスチンが正常化しても、局所的な網膜組織の酸化ストレスは進行し続けることがあります。生後3ヶ月で治療開始しても網膜変性が進むケースが報告されています。
誤解③「遅発型=軽症」
遅発型cblC病は精神症状・脊髄症・腎不全・肺塞栓症など生命に関わる多彩な症状を呈します。「乳児期に発症しなかったから軽症」という誤解は診断を遅らせる大きな要因です。
誤解④「MMACHCが陰性なら違う病気」
MMACHCのDNA配列が正常でも、PRDX1変異によるepi-cblCが存在します。臨床的にcblC病が強く疑われる場合、メチル化解析とPRDX1遺伝子検査まで行う必要があります。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 コバラミン代謝異常症・遺伝子検査のご相談
cblC病をはじめとする希少遺伝性代謝疾患の遺伝子検査・遺伝カウンセリングは、
臨床遺伝専門医が在籍するミネルバクリニックにご相談ください。
関連記事
参考文献
- [1] MedlinePlus Genetics. MMACHC gene. National Library of Medicine. [MedlinePlus]
- [2] OMIM #277400. Methylmalonic aciduria and homocystinuria, cblC type. Johns Hopkins University. [OMIM]
- [3] Carrillo-Carrasco N, et al. Combined methylmalonic acidemia and homocystinuria, cblC type. I. Clinical presentations, diagnosis and management. J Inherit Metab Dis. 2012;35(1):91-102. [PMC4219318]
- [4] Carrillo-Carrasco N, et al. Combined methylmalonic acidemia and homocystinuria, cblC type. II. Complications, pathophysiology, and outcomes. J Inherit Metab Dis. 2012;35(1):103-114. [PMC3529128]
- [5] Lerner-Ellis JP, et al. Spectrum of mutations in MMACHC, allelic expression, and evidence for genotype-phenotype correlations. Hum Mutat. 2009;30(7):1072-1081. [PubMed]
- [6] Guéant JL, et al. PRDX1 gene-related epi-cblC disease is a common type of inborn error of cobalamin metabolism with mono- or bi-allelic MMACHC epimutations. Clin Epigenetics. 2021;13(1):137. [PMC8254308]
- [7] Clinical and genetic characteristics of late-onset cobalamin C deficiency: A multicenter study in northern China. PMC. 2025. [PMC12672148]
- [8] Ruiz-Mercado M, et al. Structural Insights into the MMACHC-MMADHC Protein Complex Involved in Vitamin B12 Trafficking. J Biol Chem. 2016;291(6):2784-2794. [PMC4705923]
- [9] An unusual Co–S bond links B12 chaperones in an interprotein complex. PNAS. 2025. [PNAS]
- [10] Dong R, et al. Experimental insights into MMACHC variants using a novel plasmid system. PubMed. 2025. [PubMed]
- [11] Retinal Changes in Early-Onset cblC Methylmalonic Acidemia. Genes (Basel). 2025;16(6):635. [MDPI]
- [12] Milder Form of Cobalamin C Disease May Be Missed by Newborn Screening: The Importance of Methylmalonic Acid Assessment. Int J Neonatal Screen. 2025;11(3):77. [MDPI]
- [13] Analysis of hydroxocobalamin dosage in patients with CblC deficiency. PMC. 2025. [PMC12369215]
- [14] ClinicalTrials.gov. EPI-743 in Cobalamin C Defect (NCT01793090). [ClinicalTrials.gov]
- [15] PTC Therapeutics. FDA Acceptance and Priority Review for Vatiquinone NDA. [IR PTC]
- [16] Open Label Study of mRNA-3704 in Patients With Isolated Methylmalonic Acidemia. Boston Children’s Hospital. [BCH]






