目次
遺伝形式とは当該形質が子孫に受け渡されて性質を発現する形式のことです。常染色体優性遺伝・常染色体劣性遺伝・X関連優性遺伝・X関連劣性遺伝・ミトコンドリア遺伝・多因子遺伝に加え最新の概念である2遺伝子遺伝も説明しています。
遺伝形式とは?
遺伝形式とは、遺伝子がどのように世代から世代へと受け継がれるかのパターンを指し、生物の特徴や病気がどのように親から子へ遺伝するかを説明するために用います。ここで主な遺伝形式として、以下を挙げることができます:
1. 単一遺伝子遺伝(メンデル遺伝):
– 優性遺伝:一方の遺伝子が他方を支配し、表現型に影響を与えるパターン。
– 劣性遺伝:特定の形質が現れるためには、対になる遺伝子が両方とも劣性である必要がある。
– 共優性遺伝:二つの異なる優性遺伝子が共に影響を及ぼし、新たな表現型を作り出す場合。
2. X連鎖遺伝:
特定の遺伝子がX染色体上に存在し、性別によって表現型が異なるパターンです。
3. ミトコンドリア遺伝:
遺伝情報がミトコンドリア内のDNAから来るもので、主に母親から子へと遺伝します。
4. 多因子遺伝:
複数の遺伝子と環境要因が組み合わさって特定の形質や疾患が発現する遺伝形式。例えば、糖尿病や心臓病などの生活習慣病は、多因子遺伝の影響を受ける可能性が高いです。
5. 2遺伝子遺伝:
2つの異なる遺伝子が特定の形質や病気の発現に直接的に関与するパターン。これにより、単一遺伝子遺伝と異なり、形質の表現が複雑化することがあります。例として、ある種の遺伝的疾患が2遺伝子遺伝によって引き起こされることがあります。
これらの遺伝形式は、遺伝学の研究や遺伝病の診断、治療戦略の策定において重要です。
メンデル遺伝とは?
古典的なメンデル遺伝には常染色体優性、常染色体劣性、X染色体連鎖、ミトコンドリア遺伝があります。
遺伝の基本的な法則は、病気の伝播パターンを理解する上で大変重要となります。
ある家族が病気にかかった場合、正確な家族歴が疾患伝達のパターンを確立するために重要になりますが、昨今の核家族化で難易度が増しています。
さらに、家族歴は、特に行動や環境が強い役割を果たす一般的な病気の場合には、遺伝性疾患を除外するのにも役立ちます。
メンデルの法則
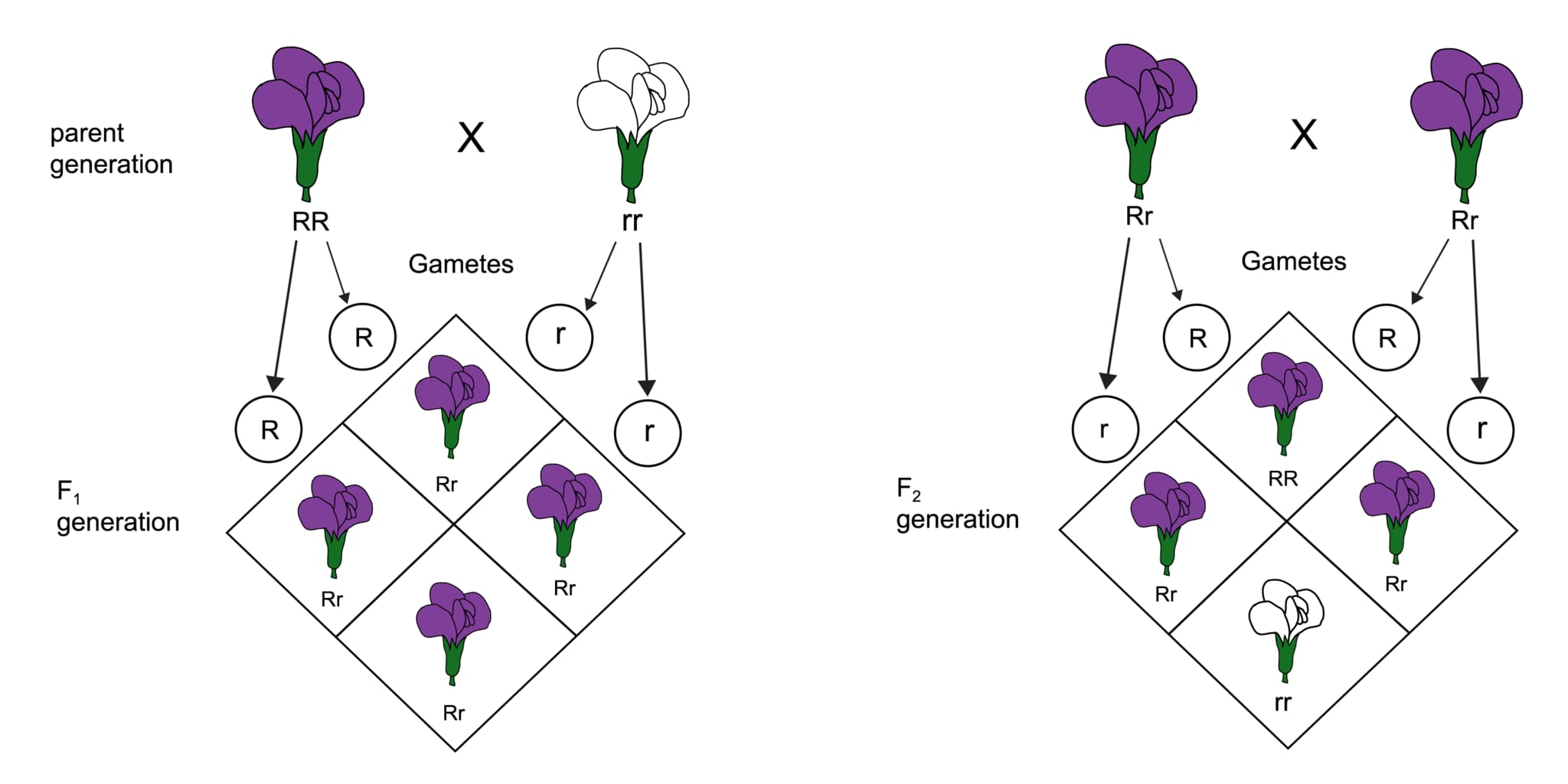
メンデル遺伝は、オーストリアの修道士グレゴール・ヨハン・メンデルが発見した遺伝の基本法則に基づく遺伝の形式です。メンデルはエンドウ豆を使った実験を通じて、遺伝子がどのように世代間で伝わるかの規則性を解明しました。彼の発見から導かれる法則は「メンデルの法則」と呼ばれ、単純な形質の遺伝パターンを説明するのに使用されます。以下はメンデル遺伝の主要な三つの法則です。
1. 分離の法則(第一法則):
この法則は、各個体が親から受け継ぐ遺伝子(対立遺伝子)が生殖細胞を形成する際に分離し、それぞれの生殖細胞には片方の対立遺伝子のみが含まれると説明しています。この結果、遺伝子のどちらが次世代に伝わるかは偶然に依存します。
2. 独立の法則(第二法則):
独立の法則は、異なる遺伝子対が互いに独立して遺伝するというものです。つまり、ある特定の遺伝子の遺伝が他の遺伝子の遺伝に影響を与えることはありません(ただし、これは遺伝子の位置が異なる染色体上にある場合に限ります。同一染色体上の遺伝子では連鎖という現象が発生します)。
3. 優性の法則(優性と劣性の法則):
優性の法則によると、対立遺伝子の組み合わせにおいて一方が他方を支配(優性)する場合、優性遺伝子の形質のみが表現型として現れます。劣性の遺伝子が表現型として現れるには、その遺伝子が両親から受け継がれる必要があります。
メンデル遺伝は、生物学や遺伝学の基礎を理解するうえで非常に重要な概念であり、多くの生物の形質がどのように遺伝するかを説明する際に用いられます。
単一遺伝子疾患
単一遺伝子疾患(単因子遺伝病)とは、一つの遺伝子の変異によって引き起こされる遺伝性の疾患です。これらの病気は、特定の遺伝子に発生した変異が直接的に病態を形成するため、その遺伝パターンはメンデル遺伝の法則に従うことが多いです。単一遺伝子疾患は、その遺伝形式によって優性遺伝、劣性遺伝、X連鎖遺伝、ミトコンドリア遺伝に分類されます。
ほとんどの遺伝子は、対立遺伝子と呼ばれる突然変異や多型により、1つ以上のバージョンを持っています。
個人は、突然変異や多型の影響とその対立遺伝子の集団における頻度に応じて、「正常」対立遺伝子、「病的」または「稀な」対立遺伝子を保有している可能性があります。
単一遺伝子疾患は、通常、遺伝子の位置および疾患表現型を発現させるために遺伝子の正常コピーが1つまたは2つ必要かどうかに応じて、いくつかのパターンのうちの1つで遺伝します。
正常対立遺伝子に対する突然変異対立遺伝子の発現は、優性、優性、劣性、または劣性として特徴づけられます。
単一遺伝子疾患の基本的な遺伝様式は、常染色体優性、常染色体劣性、X連鎖陽性、X連鎖劣性、ミトコンドリアの5つとなります。
ここでいくつかの代表的な単一遺伝子疾患を挙げます:
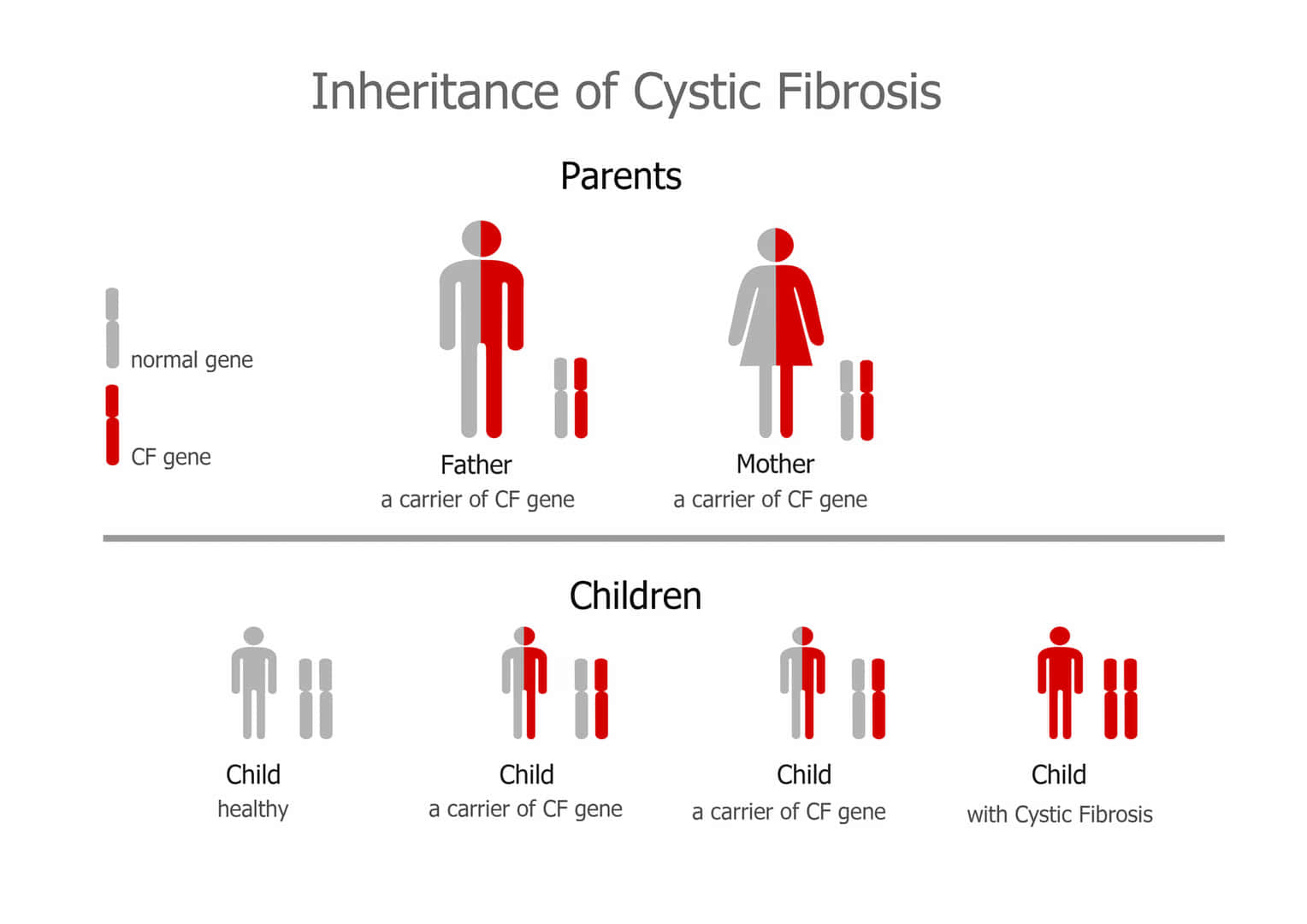
1. 嚢胞性線維症:
CFTR遺伝子の変異により引き起こされる劣性遺伝の疾患で、主に肺や消化器系の粘膜分泌異常を特徴とします。
2. ハンチントン病:
HTT遺伝子の変異による優性遺伝の疾患で、神経細胞が徐々に破壊されることにより、運動能力の障害や認知機能の低下が進行します。
3. デュシャンヌ型筋ジストロフィー:
DMD遺伝子の変異によるX連鎖劣性遺伝の疾患で、筋力の低下や筋肉の変性を引き起こします。
これらの疾患は診断、予防、治療において遺伝学的アプローチが重要であり、遺伝カウンセリングを通じて家族歴の評価やリスク評価が行われることがあります。単一遺伝子疾患の理解は、個別化医療の進展にも寄与しています。
遺伝的不均一性
遺伝的不均一性とは、同じまたは類似の症状を示すが、その原因となる遺伝子が異なる状態を指します。この現象は、特に遺伝性疾患の研究において重要です。遺伝的不均一性は、大きく二つのタイプに分けられます。
1. 遺伝子的不均一性(アレル不均一性):
同一遺伝子内の異なる変異が同じ疾患を引き起こす場合です。つまり、同じ遺伝子の異なる位置の変異が同じ形質や病気を引き起こします。例えば、嚢胞性線維症はCFTR遺伝子の異なる変異によって発症する場合があります。
2. 遺伝子間不均一性(局所不均一性):
異なる遺伝子の変異が類似の症状を引き起こす場合です。例えば、網膜色素変性症や先天性聴覚障害など、複数の異なる遺伝子の異常が同様の疾患を引き起こすことが知られています。
遺伝的不均一性の存在は、遺伝病の診断や治療の戦略に重要な影響を与えます。特定の疾患に対して遺伝子検査を行う際、一つの遺伝子だけでなく複数の遺伝子を調べる必要があることがあり、これは遺伝子パネル検査や次世代シーケンサーによる広範な遺伝子解析が求められる理由の一つです。また、遺伝的不均一性の理解は、個々の患者に最適な治療法を選択するための精密医療にも寄与します。
遺伝的不均一性は、家族内で異なる疾患の重症度や経過が観察されることがある現象です。これは、単一遺伝子疾患だけでなく、より複雑な多因子性疾患にも見られます。遺伝的不均一性には以下のような要因が関与しています。
1. 遺伝子間不均一性:上述
2. 遺伝子的不均一性:上述
3. 修飾遺伝子の影響:ある遺伝子の表現型が他の遺伝子によって修飾されること。これにより、同じ遺伝的背景を持つ個体間でも疾患の表現に大きな違いが生じることがあります。
4. 環境要因:生活習慣や環境が遺伝的要因と組み合わさり、疾患の表現型に影響を与える場合。これは特に多因子性疾患で重要な役割を果たします。
遺伝的不均一性の理解は、遺伝病の正確な診断と効果的な治療戦略の策定に不可欠です。家族歴の詳細な評価や遺伝子検査を通じて、個々の患者に最適な治療アプローチを決定するための重要な情報が得られるため、精密医療の推進に寄与しています。
単一遺伝子疾患に関する情報は、
Online Mendelian Inheritance in Man (OMIM; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM)
GeneTests/GeneClinics (http://www.genetests.org)から得ることができます。
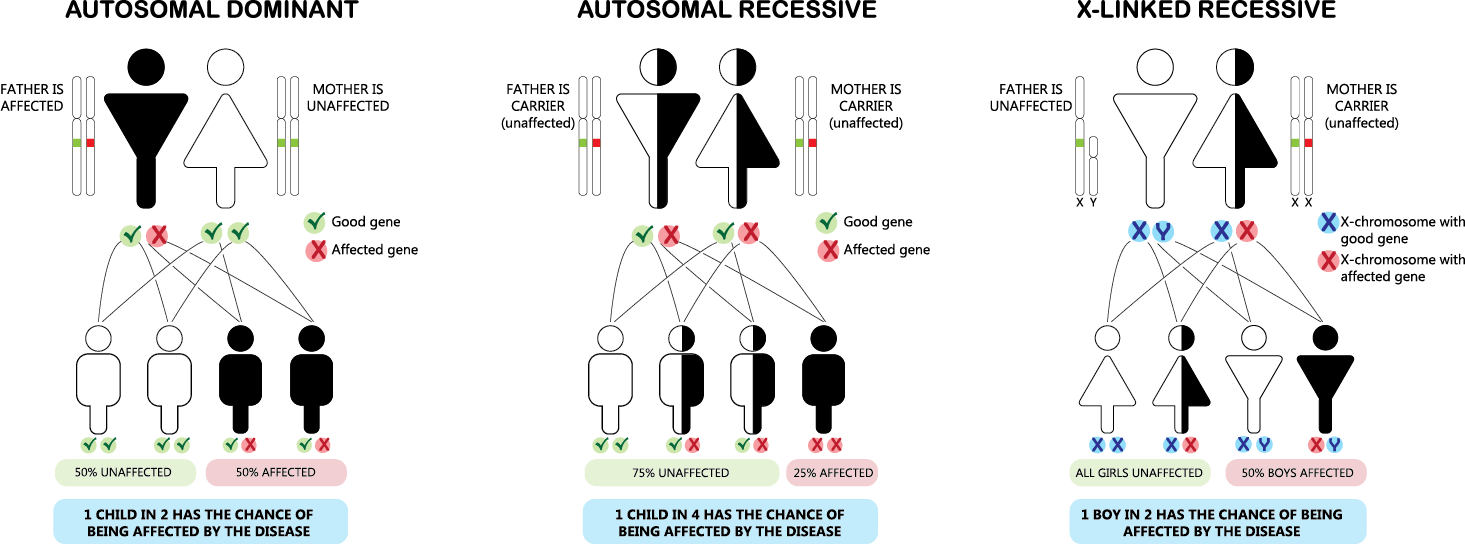
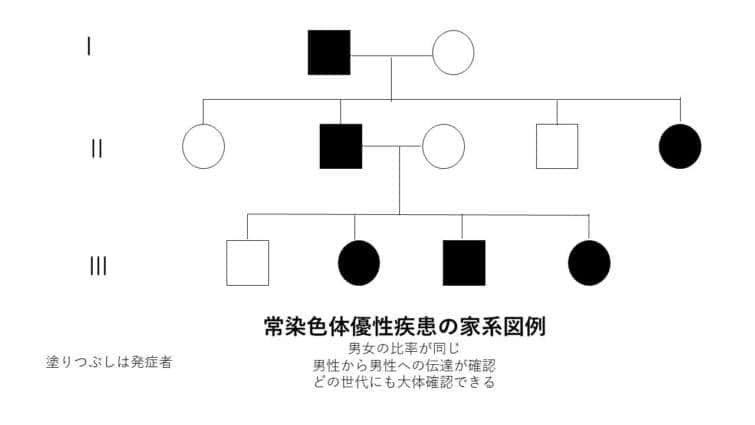
常染色体優性遺伝
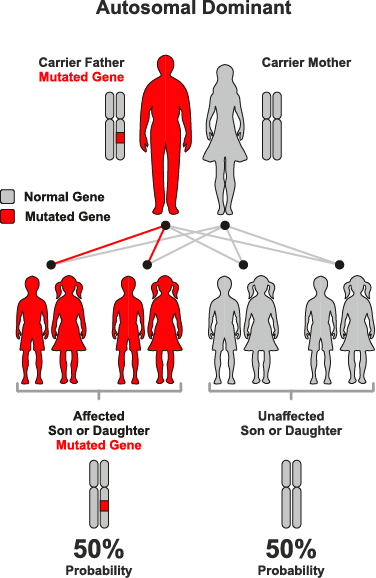
常染色体優性遺伝は、遺伝形式の一つで、特定の遺伝子の変異が常染色体(性染色体以外の染色体)のうちの一方に存在するだけで、表現型として症状が現れる場合を指します。この遺伝のパターンでは、変異を持つ一つの遺伝子が正常な遺伝子を支配する(優性)ため、病気や特定の形質が発現します。
● 常染色体優性遺伝の特徴:
– 片親からの遺伝: 変異を持つ片親から子へ遺伝する可能性が50%あります。変異遺伝子を持たない親からはこの病気を引き継ぐことはありません。
– 性別に依存しない: 常染色体優性遺伝は男女の区別なく発現します。
– 代を越えた症状の現れ方: 症状は世代を通じて連続して現れる傾向があり、片親が症状を持つ場合、その子どもの半数が症状を持つ可能性があります。
● 例:
1. ハンチントン病: 神経変性を引き起こす代表的な常染色体優性遺伝疾患です。特定の遺伝子(HTT遺伝子)の特定の領域が異常に繰り返されることで発症します。
2. 骨形成不全症: 脆弱な骨が特徴的な病気で、COL1A1またはCOL1A2遺伝子の変異により起こります。
常染色体優性遺伝に関連する疾患の診断と治療は、遺伝カウンセリングや遺伝子検査を通じて行われることが一般的です。これにより、家族内でのリスク評価や将来的な健康管理の計画が可能になります。また、この遺伝形式の理解は、遺伝病の研究や新たな治療法の開発にも寄与しています。
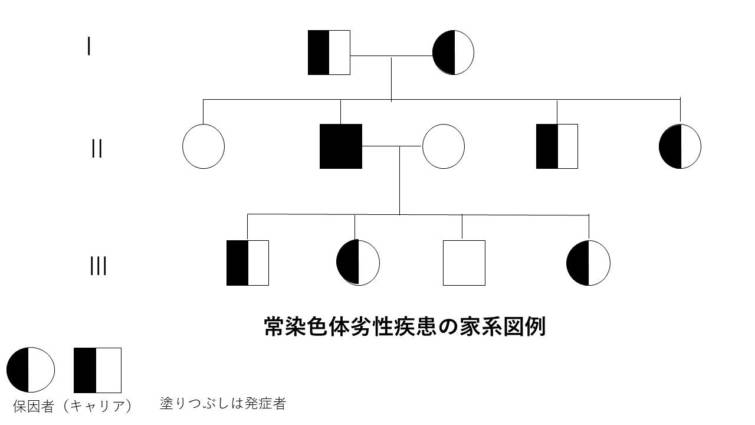
常染色体劣性遺伝
常染色体劣性遺伝は、特定の遺伝子の変異が常染色体(性染色体以外の染色体)において両方に存在する場合にのみ表現型として症状が現れる遺伝のパターンです。この場合、一方の遺伝子だけが変異している状態(ヘテロ接合体)では通常、症状は現れません。症状が表れるためには、両方の遺伝子が変異している必要があります(ホモ接合体)。
● 常染色体劣性遺伝の特徴:
– 両親からの遺伝: 症状を示すためには、両親からそれぞれ変異遺伝子を受け継ぐ必要があります。通常、両親は変異遺伝子の保因者(キャリア)であり、症状は示さないことが多いです。
– 性別に依存しない: 常染色体劣性遺伝は男女に等しく影響します。
– 保因者の存在: 保因者(変異遺伝子を一つだけ持つヘテロ接合体)は一般に症状を示しませんが、特定の疾患では保因者にも軽度の症状が見られることがあります。
● 例:
1. 鎌状赤血球病: ヘモグロビンの遺伝子に変異があり、赤血球が特異的なシックル(鎌)形を呈することで知られています。両親から変異遺伝子を受け継いだ場合に症状が現れます。
2. 嚢胞性線維症: CFTR遺伝子の変異により引き起こされる病気で、主に肺や消化器系の粘膜分泌異常を特徴とします。両親から変異遺伝子を受け継ぐことにより症状が表れます。
常染色体劣性遺伝疾患の診断には遺伝子検査が不可欠であり、遺伝カウンセリングを通じて家族内のリスクの評価や健康管理が行われます。また、保因者の特定は家族計画において重要な情報となり得ます。この遺伝形式の理解は、遺伝病の研究だけでなく、適切な治療や予防戦略の策定にも寄与しています。
常染色体劣性疾患の多くは、同一祖先の子孫であるため、同一遺伝子の突然変異を保有している可能性が高いことから、血縁関係にある人に多く見られます。多くの常染色体劣性疾患は、これらの個体が創始者とよばれる同じ祖先の子孫であるため、特定の民族背景を持つ個体に他の個体よりも多く見られます。しかし、これらの共通の祖先は、一般的にこれらの個人とより遠縁であるため、同じ民族的背景を持つカップルは、一般的に血縁関係にあるカップルよりも共通の遺伝子の数が少ないことになります。
X連鎖優性遺伝
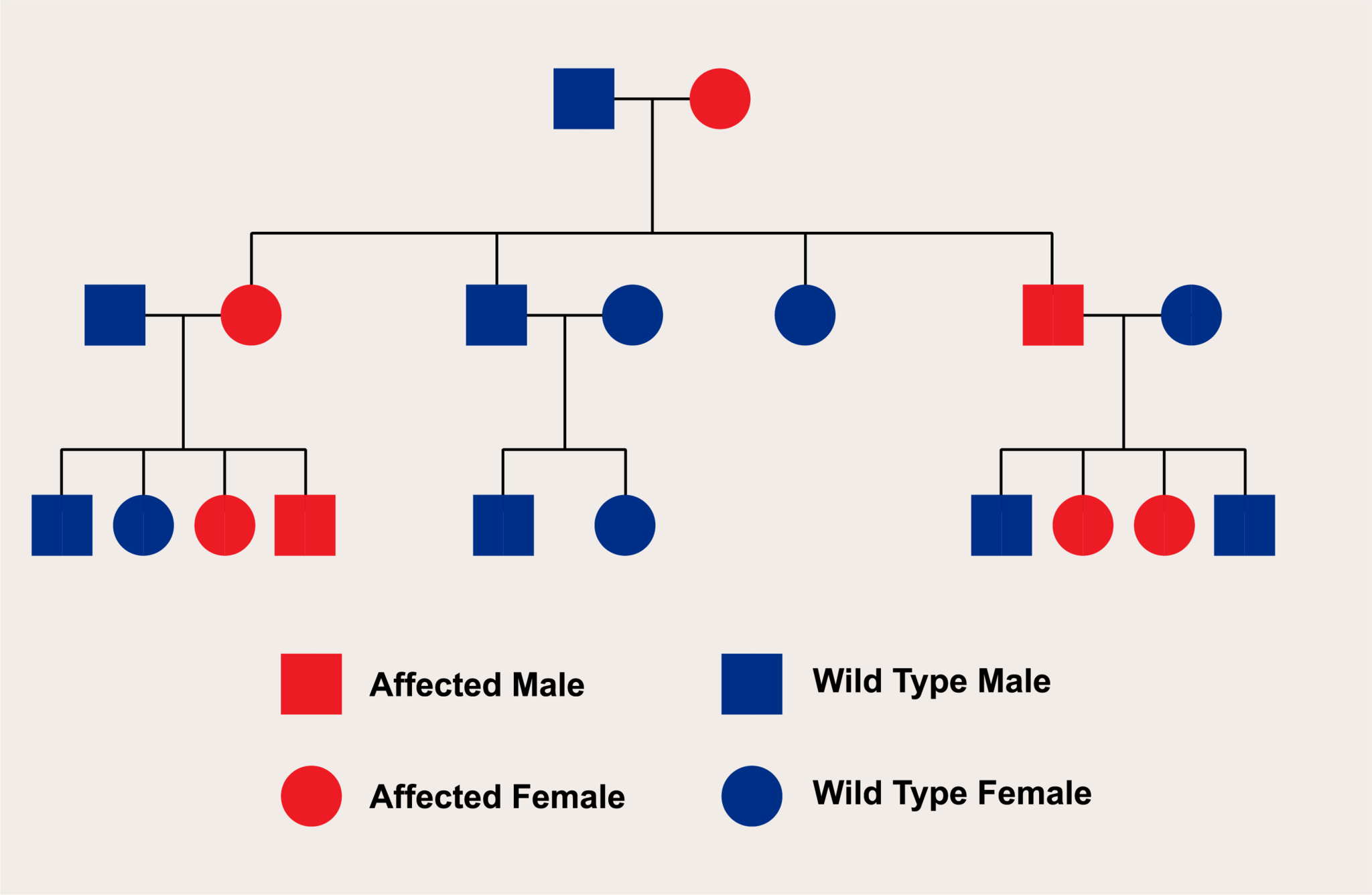
X連鎖優性遺伝は、特定の遺伝子の変異がX染色体上に存在し、その変異が優性の特性を持つ遺伝のパターンを指します。このタイプの遺伝では、変異がX染色体にあるため、遺伝の動態が性別によって異なる影響を受けます。
● X連鎖優性遺伝の特徴:
1. 女性における遺伝:
女性はX染色体を二つ持っているため、変異遺伝子を一つだけ持つヘテロ接合体の状態でも症状が表れる可能性があります。ただし、優性遺伝であっても、表現の程度には個体差があることがあります。
2. 男性における遺伝:
男性はX染色体を一つしか持たないため、変異遺伝子を持つと、その遺伝子の影響が直接表れます。そのため、男性は変異遺伝子を持つとほぼ確実に症状が現れます。
3. 遺伝のパターン:
変異遺伝子を持つ女性(ヘテロ接合体またはホモ接合体)は、その子供に変異遺伝子を伝える確率が50%です。男性が変異遺伝子を持つ場合、彼の全ての娘がその遺伝子を受け継ぎますが、息子には伝えません。これは、男性がY染色体のみを息子に伝えるためです。
● 例:
– ビタミンD依存性くる病タイプI (VDDR-I):この疾患は、1α-ヒドロキシラーゼ(CYP27B1)遺伝子の変異により引き起こされ、X連鎖優性のパターンを示します。これにより、カルシウムとリンの代謝が乱れ、骨の形成に問題が生じます。
X連鎖優性遺伝疾患の理解は、診断、家族計画、および遺伝カウンセリングにおいて重要です。特に、性別による症状の違いや遺伝のリスクを家族に説明することは、遺伝性疾患の管理において非常に重要な要素となります。
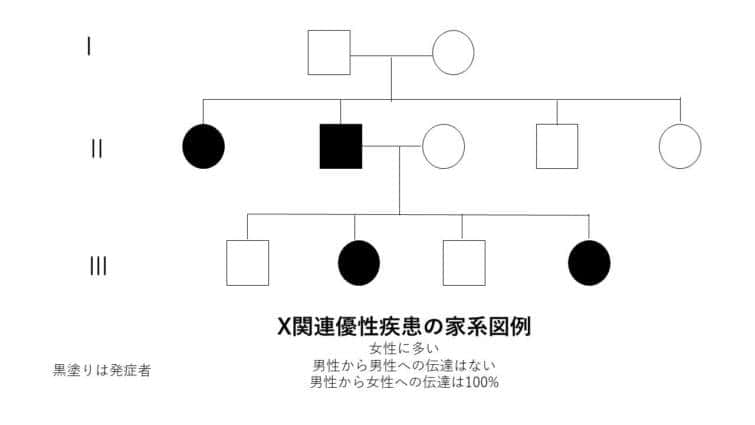
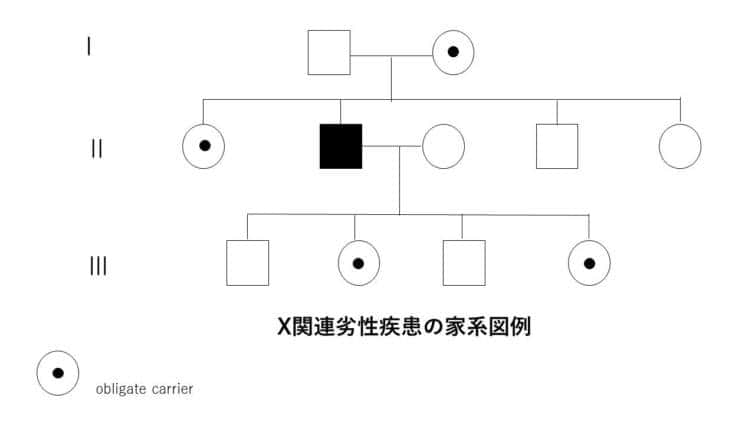
X連鎖劣性遺伝
X連鎖劣性遺伝は、特定の遺伝子の変異がX染色体上に存在し、その変異が劣性の特性を持つ遺伝のパターンを指します。このタイプの遺伝では、変異がX染色体にあるため、性別によってその影響が大きく異なります。
● X連鎖劣性遺伝の特徴:
1. 男性における遺伝:
男性はX染色体を一つしか持っていないため、変異遺伝子を受け継いだ場合、劣性遺伝子であってもその影響が直接表れます。これは、男性がもう一つの健康なX染色体を持っていないため、変異が即座に表現型として現れることを意味します。
2. 女性における遺伝:
女性はX染色体を二つ持っており、変異遺伝子が一つしかない場合(ヘテロ接合体)は、もう一方の健康なX染色体が変異の影響を相殺することが多いです。そのため、通常は症状が表れませんが、保因者として変異を次世代に伝える可能性があります。
3. 遺伝のパターン:
変異遺伝子を持つ女性(保因者)は、その子供に変異遺伝子を伝える確率が50%です。男性が変異遺伝子を持つ場合、全ての娘が保因者になる一方で、息子には変異遺伝子は伝わりません。
● 例:
– デュシェンヌ型筋ジストロフィー:
DMD遺伝子の変異により引き起こされるX連鎖劣性遺伝病で、この遺伝子は筋肉の構造と機能に重要な役割を果たします。男性患者では筋力の低下や筋肉の変性が見られ、進行すると生命を脅かすこともあります。
– 色覚異常:
色を識別する能力に影響を与える遺伝病で、特に赤緑色盲がよく知られています。保因者の女性は通常、症状がないですが、男性では一般に色覚異常の症状が顕著に現れます。
X連鎖劣性遺伝疾患の理解は、遺伝カウンセリングや家族計画において重要です。保因者である女性が自身の遺伝的状態を知ることは、子供に病気が遺伝するリスクを評価する上で不可欠です。この情報は、適切な医療対策や予防策を計画する際にも役立ちます。
X連鎖劣性遺伝の血友病が女性で発症するケースは比較的まれですが、いくつかの特定の状況下で起こり得ます。女性が血友病の症状を示す主な理由は以下のとおりです。
1. ホモ接合体女性:
女性がX染色体上の血友病関連遺伝子(例:F8遺伝子やF9遺伝子)の変異を両方のX染色体上で持っている場合、彼女はホモ接合体であり、血友病の症状が現れます。この状態は非常に稀で、通常は両親が共に変異のキャリアである、または片親が血友病患者であり、もう片親が変異のキャリアである場合に生じます。
2. 重複X染色体欠失:
女性が片方のX染色体に欠損や異常を持ち、もう片方のX染色体が血友病の変異を持っている場合、健康なX染色体が不活性化されるか機能しないため、血友病の症状が現れることがあります。
3. X染色体の不活性化の不均一性(ライオニゼーション):
通常、女性は二つのX染色体を持っていますが、体細胞の中で一方のX染色体は無作為に不活性化されます(X-インアクティベーション)。しかし、この不活性化が不均一である場合(すなわち、健康なX染色体が不活性化され、変異を持つX染色体が活性化される確率が高い場合)、血友病の表現型が現れる可能性があります。
これらの特殊なケースにより、X連鎖劣性の遺伝病が女性にも発現することがあります。血友病のような疾患は、遺伝カウンセリングや遺伝子検査を通じて、個々の遺伝的リスクを評価し、適切な治療や管理を行うことが重要です。
ミトコンドリア遺伝
ミトコンドリアは、エネルギー生産を担当しています。ミトコンドリアは、滑らかで連続的な外膜と、管状またはひだ状に配置された板状の二重膜(クリスタエ)の2つの膜から構成されています。ミトコンドリアは、実際には細胞の主要なエネルギー源です(末端電子輸送のチトクロム酵素とクエン酸サイクル、脂肪酸酸化、酸化的リン酸化の酵素のおかげで)。ミトコンドリアは、栄養素をエネルギーに変換するだけでなく、他の多くの専門的な働きをしています。
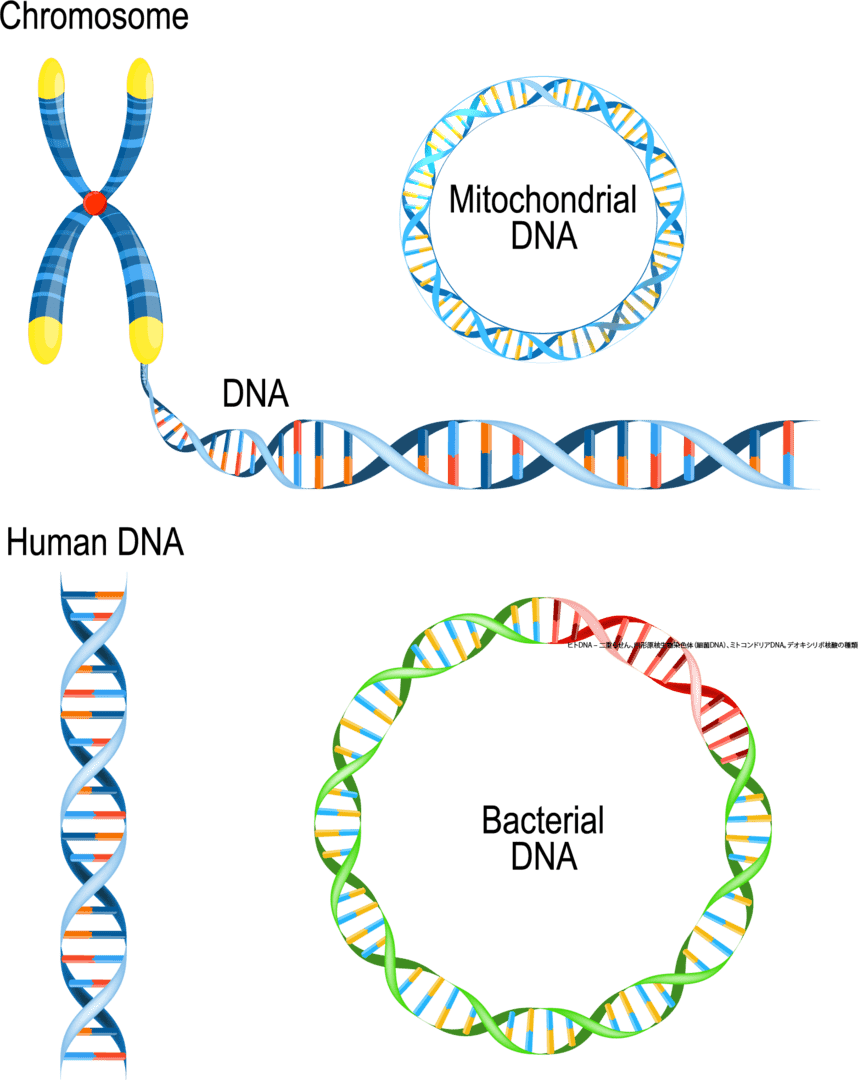
各ミトコンドリアは、DNA(ミトコンドリアDNAまたはmtDNA)でできた染色体を持っていますが、核内のよく知られた染色質とは全く異なります。ミトコンドリア染色体ははるかに小さいです。核内の染色体が棒状であるのに対し、ミトコンドリア染色体は丸い形をしています。どの細胞にもミトコンドリア染色体のコピーがたくさんあります(通常、核には1セットの染色体しかありません)。ミトコンドリアのDNAには37個の遺伝子が含まれていますが、これらはすべてミトコンドリアの正常な機能に不可欠なものです。多くの遺伝的疾患が、特定のミトコンドリア遺伝子の変化に関連しています。
ミトコンドリア遺伝とは、ミトコンドリアゲノムにコードされた形質が継承されることをいいます。
ミトコンドリアは、細胞内の正常な構造物または小器官です。ミトコンドリアは、核の外側の細胞質に位置しているため、ミトコンドリア遺伝は体細胞遺伝ともいわれます。
ミトコンドリアは、末端電子輸送のチトクロム酵素とクエン酸サイクル、脂肪酸酸化、酸化的リン酸化の酵素を内包し、エネルギー生産を担当しています。ミトコンドリアは、滑らかで連続的な外膜と、管状またはひだ状に配置された板状の二重膜で構成されています。
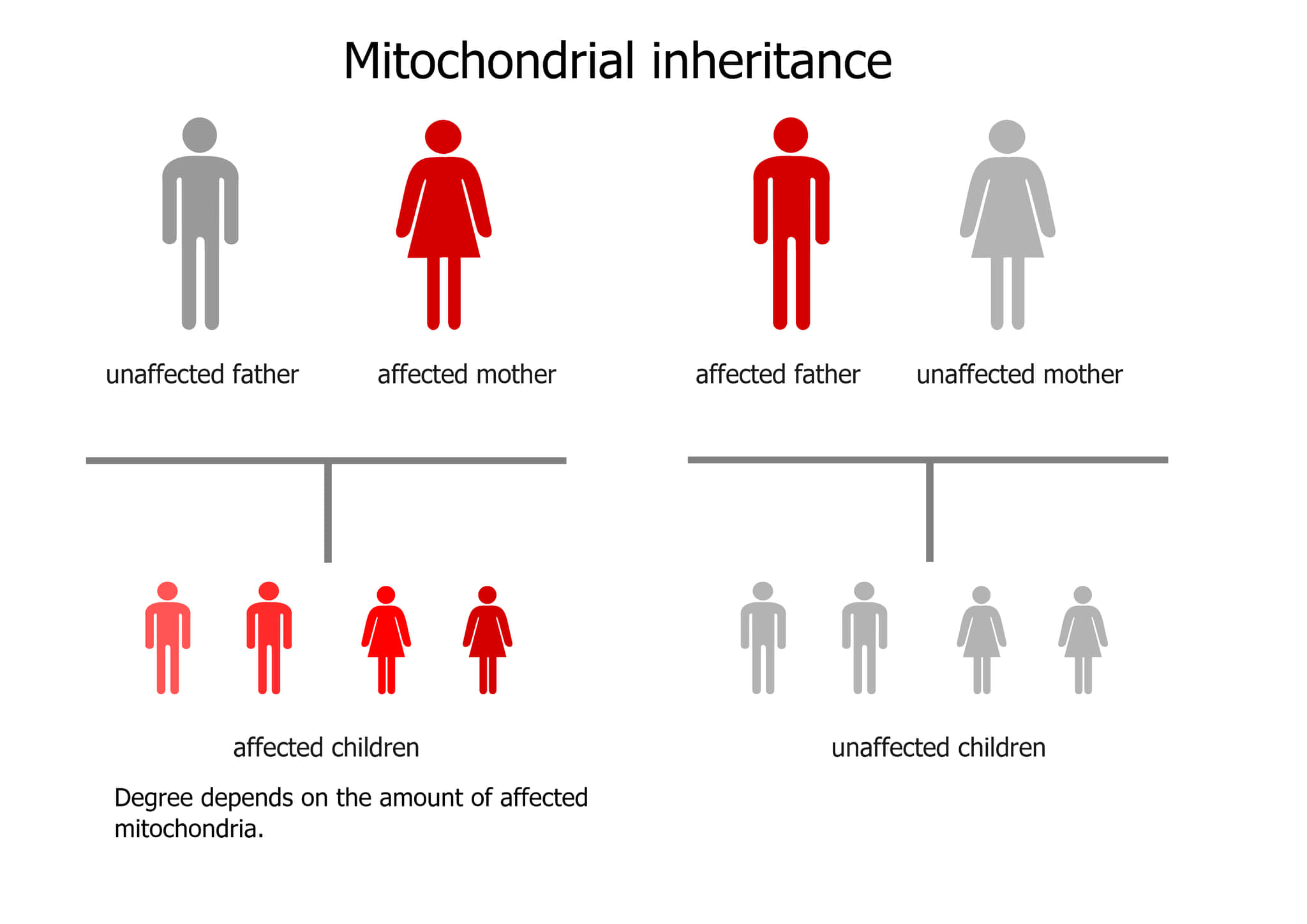
ミトコンドリア遺伝は母系遺伝である
ほとんどの多細胞生物では、mtDNAは母親から遺伝するので母系遺伝です。
ミトコンドリア遺伝が母系遺伝であるメカニズムには以下の二つがあります。
- 1.ヒトの卵子には平均20万個のmtDNA分子が含まれている
- 2.ヒトの精子には平均5個の分子が含まれていることが報告されている
- 3.男性生殖管内および受精卵内での精子のmtDNAの分解
- 4.少なくともいくつかの生物では、精子のmtDNAが卵子に入ることができないことが報告されていてヒトでもそういう機序がある可能性がある
精子のでき方についてはこちらをクリックしてご覧ください
関連記事:精子形成
どのようなメカニズムであれ、この片親(片親継承)パターンのmtDNA継承は、ほとんどの動物、ほとんどの植物、そして真菌にも見られます。
ミトコンドリアのボトルネック
単親遺伝で組換えがほとんどない個体は、ミュラーのラチェット、すなわち機能が失われるまでの間に有害な突然変異が蓄積されることが予想されます。
ミトコンドリアの恩恵にあずかる動物個体群は、mtDNAボトルネックとして知られている発生過程を経て、これを回避しています。
ちなみに、なぜ細胞内小器官でミトコンドリアだけがミトコンドリアゲノムと呼ばれる独自のDNAを持つのか不思議じゃないですか?実は、ミトコンドリアは太古はヒトと別の生物(好気性細菌:糖や脂質のような基質を酸化してエネルギーを得る)だったのが、ヒトに感染して寄生しているのです。好気性細菌のミトコンドリアは他の細菌と同じく宿主がいないと生きられず、また、ヒトにとってはエネルギーを産生してくれるという大変好都合な細菌だったため、普通は感染すると免疫力により排除されるのですが、ミトコンドリアとヒトはお互い一緒にいることが大きなメリットなのでラブラブでそれ以来ずっと細胞のなかで共生しているということです。
生物は細胞の中に核があるかないかで核がない原核生物、核がある真核生物です2分できます。原核生物である細菌には核がないので細胞質の中にDNAがそのままあり、を持っていますが、核のようにDNAの周りを隔てる仕切りはありません。DNAがある程度まとまって細胞の中にあり、環状の遺伝子のことが多いです。ミトコンドリアの遺伝子も環状になっていて、これがミトコンドリア寄生体説の裏付けになっています。
話を元に戻しましょう。
ボトルネックってなあに?ってことですが。
実は細胞の中のミトコンドリアは1種類ではありません。いや。ミトコンドリアとしては1種類なのですが、たとえば100人のヒトが生物種としてはヒトと分類されても一人一人は別であるように、ミトコンドリアも遺伝子レベルで分類するとそれぞれたとえばABCDEFG・・・・という風に複数の種類のミトコンドリアが細胞内にはあるのです。その割合も臓器や組織の単位でバラバラです。たとえば心臓ではABCがそれぞれ2:5:3の割合であるが、骨格筋ではCDEが7:2:1という風に。
そして、このうちたとえばAに突然変異が起こり機能不全になったとします。
ミトコンドリア病がややこしいのは、Aに機能不全がおこってもBCの正常な系統が多ければ細胞全体として機能するため別に問題はない=症状がでない、ということです。そして、受け継がれていく過程で突然Aがたとえば80%とかになってしまうとほかのBCが一生懸命働いても補いきれなくなり、ミトコンドリアがその割合を示す臓器でのみ表現型(病気)が発現する、ということです。
ミトコンドリアのボトルネック現象とは、卵子形成過程で一度ミトコンドリアの数が劇的に少なくなった後にミトコンドリアの分裂により元の数にもどる現象をいいます。真核生物はこの過程により機能異常をもつミトコンドリアを淘汰して自分の生存に都合の良いミトコンドリアを選択していると考えられています。これとは全く逆に、ボトルネックの過程をへるときに、異常なミトコンドリアが選択されてしまい、その比率が急に増えて、お母さんは正常なのにお子さんが急に発症することもあり、なかなか読めないのがミトコンドリア遺伝です。
39組の母子のmtDNAの研究からは、ほとんどの母子で、mtDNAにバリアントがあり、変異の1/8は病的ではないが病気を起こす可能性がある(likely pathogenic)バリアントでした。しかし、アミノ酸変異を起こすタイプのバリアントはボトルネックの際淘汰されるのか伝達されにくいこともわかりましたが、母体の年齢が20代にと30代とで比較するとmtDNA異常の伝達率は2−3倍になることもわかりました。そして、1/39で子供の細胞でバリアントmtDNAが急増していたと報告されています。生殖細胞におけるボトルネックはこのようにミトコンドリア遺伝に複雑性を提供しています。ボトルネックでミトコンドリアは10万以上から一気に40以下に減少する事も明らかにされています。
要するにボトルネックは細胞内のランダムなプロセスを利用して、生物の発生に伴って突然変異負荷の細胞間のばらつきを増大させ、その後、細胞レベルでの選択が作用して、より多くの突然変異mtDNAを持つ細胞を除去し、世代間での突然変異負荷の安定化または減少に寄与している可能性があるといえます。
ボトルネックの根底にあるメカニズムは議論の途上ですが、最近の数学的および実験的メタスタディーにより、細胞分裂時のmtDNAのランダムな分割と細胞内でのmtDNA分子のランダムなターンオーバーの組み合わせがボトルネック現象のメカニズムであるとの証拠が提供されています(こちらをクリックしてご覧ください)。
ミトコンドリアのヘテロプラスミー
ミトコンドリアの遺伝の理解を難しくしている原因が、ヘテロプラスミーと呼ばれる現象で、一つの細胞に正常と突然変異を持ったミトコンドリアが共存することを言います。ミトコンドリアゲノムが独立性を持つため起こる状態なのですが、一つのミトコンドリアで突然変異が起こって機能不全となっても、細胞内には多くの正常ミトコンドリアが存在して機能不全におちいったミトコンドリアの機能を代償するため異常が表に出ません。しかし、分裂しない神経細胞などでは異常ミトコンドリアの増殖が高まってしまい、割合が閾値を超えてしまうと症状が発現することとなります。
ヘテロプラスミーとは、細胞または個体内に2種類以上のmtDNAが存在することをいいます。これは、ミトコンドリア疾患の重症度を考慮する上で非常に重要な因子となります。ほとんどの真核細胞には数百のミトコンドリアがあり、ミトコンドリアDNAの数百のコピーがあるため、突然変異がおこっても一部のミトコンドリアだけに影響を与え、ほとんどのミトコンドリアは影響を受けないのが一般的なのです。
逆に、細胞の中のミトコンドリアDNAが一種類である時(正常の場合も変異の場合もある)は、ホモプラスミーと呼んでいます。
ヘテロプラスミーの程度が個々の細胞ごとに異なっていることが、患者さんの全身のいろんな部位、たとえば骨格筋、血管、神経など研究から明らかとなっています。要するにミトコンドリア異常が表現(症状として表れている)されている細胞群(器官・臓器、たとえば肝臓)では変異ミトコンドリアDNAの比率が高く、それ以外の細胞(正常に働いているようにみえる細胞)ではその比率が低いため症状として表れない(表現されない)のです。
また、低い比率の変異ミトコンドリアDNAをもっていても他の大多数のミトコンドリアが正常に機能するため当該細胞の機能自体は全く持って正常であることがも明らかにされています。変異ミトコンドリアDNAをもっていること自体が必ずしも病気になることとは同義でなく、また、たとえばミトコンドリア疾患を疑ったとしても、採取した細胞群だけで正常で実はほかの細胞群には異常があったなどということで診断自体に苦慮することにもつながっています。現在、ミトコンドリアDNA検査は、ヘテロプラスミーを調べることはできず、検査に提出された細胞群全体の平均値をみているだけといえるのです。
多因子遺伝
たとえば冠動脈疾患は世界的に有病率が増加しており、昨今の先進国における死亡原因の第一位となっていますが、この疾患は部分的には遺伝によって説明されており、家族内で発症することが知られています
(Genetic susceptibility to coronary artery disease: from promise to progress Watkins et al, 2006)。
しかし、食生活などの生活習慣や環境因子も冠動脈疾患や心不全に寄与している。このように、この疾患は遺伝的要因と環境要因の両方が発症の引き金となるため、多因子遺伝を示しています。多因子とは原因が複数あるということを意味しています。
しかし、冠動脈疾患と他の多因子疾患のちがいは何なのでしょうか?
時間の経過とともに、この疑問に対する答えがどんどんと明らかになってきています。
多因子遺伝の研究のはじまり
多因子遺伝を研究した最初の科学者は、チャールズ・ダーウィンのいとこであるフランシス・ガルトンだというとても古い話です。ですので、多因子遺伝は古くて新しい遺伝概念なのです。
同時代のグレゴール・メンデルと同様に、ガルトンは形質の継承を研究しました。
しかし、メンデルとは異なり、ガルトンは「融合」と呼ばれる特徴を観察しました(Galton, 1897)。
例えば白い花の雌と赤い花の雄を交配するとピンクの花ができることを観察し、雌雄が子孫に同等の遺伝学的寄与をする現象を観察しました。
融合(ブレンディング)とは、現在では連続的変異として知られており、身長などの表現型が一つのカテゴリーに分類されない表現のグラデーションを意味します。連続的変動を示す形質をグラフにプロットすると、表現型分布はベル型(正規分布)の曲線を形成します。したがって、ほとんどの個体は中間的な表現型を持ち、大多数の個体は平均値に集まります(Mossey, 1999)。連続的な変動を持つ形質は、量的形質とも呼ばれます。このような形質では、遺伝的・環境的要因が幅広く関与しているため、多様な遺伝子型が産生されるのです。
これに対して、多因子遺伝を持つ形質の中には、階調性がないものもあります。メンデルはこれらのいわゆる「非混合」形質を研究し、それらを明確なカテゴリーに分類しました。これらの形質は、メンデルの丸くてしわくちゃなエンドウ豆のように、ある表現型から別の表現型への突然の変化があるときに起こる不連続的な変異を示しています。ここでは、丸いエンドウとしわのあるエンドウの間に中間的な変化はなく、どちらかのカテゴリーに分類されています。不連続的な変異を示す形質のもう一つの例は、ヒトのABO血液抗原系です。
メンデル理論とガルトン理論のどちらがヒトの遺伝を正しく記述しているかをめぐって、メンデルとガルトンの研究者たちは長年議論を重ねてきました。メンデル説は、いくつかのヒトの病気や形質を正しく記述していたが、そのすべてを記述していたわけではありませんでした。同様に、ガルトニアンもすべての観察結果に適合していたわけではありませんでした。今日では、両方の理論が、様々なヒトの病気を含む様々な形質の遺伝パターンを正しく記述していることがわかっています。ある病気がメンデル型かガルトン型かという問題は当該疾患に依存しているのです。
多因子疾患の特徴とは?
多因子疾患は、明確なメンデル型や性差限定型の疾患とは区別できる特徴的な特徴の組み合わせを持っています。これらの特徴には以下のようなものがあります。
1.孤発性発症
この病気は孤発性に発症し、罹患した子供が罹患していない親から生まれてくることがあります。同じ家族内に複数の症例が存在する場合、つまり家族性がある場合がありますが、明確なメンデル遺伝のパターンはありません。
2.環境因子の影響を受ける
環境の影響は、多因子疾患のリスクを増加させたり減少させたりします。
3.性別によるリスクの高低がある
多因子疾患は、一方の性別の方が他方の性別よりも多く発症しますが、性別に限定された形質ではありません。また、発症頻度の低い性別に属する人の第一度近親は、発症リスクが高いとされています(国際放射線防護委員会、2000年)。
一卵性双生児と二卵性双生児で同調率がメンデルの法則に従わない
一卵性双生児と二卵性双生児では、メンデル的な比率が矛盾しています。同調率とは、双子の両方が特定の病気にかかる率を示す比率です。(Mossey, 1999; Griffithsら, 1999)。
特定の民族で発症率が高い
この病気は、特定の民族集団(すなわち、白人、アフリカ人、アジア人、ヒスパニックなど)でより頻繁に発生します。
発症リスクを高める因子が多数存在
冠動脈疾患の例に戻ると、肥満、II型糖尿病、高血圧、低密度リポ蛋白コレステロールの高値、さらには歯周病などの発症リスクを高める因子が多数存在することがわかっています。冠動脈疾患は家族内で発症するが、メンデル遺伝パターンを示さず、孤立して発症することもあります。
冠動脈疾患は女性よりも男性に多く発症し、そのリスクは白人やアジア人よりもアフリカ系アメリカ人の方が高くなっています。これらの特徴はすべて、冠動脈疾患を多因子疾患として分類することと矛盾しません。
多因子性疾患は連続的なものなのか、それとも不連続的なものなのか?
離散的なカテゴリーに分類される形質は不連続的と呼ばれ、表現型の勾配を示すものは連続的に分類されることを思い出してください。興味深いことに、不連続的な変異に起因する疾患の中には、連続的な変異に似た複雑な表現型を示すものが多く存在します(リファレンスはこちらをクリック)。科学者たちは、これは、病気に対する感受性を発達させるための連続的な変化の基盤があるからだと提案しています。この理論によれば、病気は、ある臨界責任閾値に達した後にのみ発症し、発現するとされています。臨界責任閾値を超えると、病気の表現型はより深刻になります。対照的に、責任閾値に達していない個人は、病気を発症することはありません。したがって、個人はこの病気に罹るか罹らないかのどちらかであり、病気を発症(表現)するかどうかは不連続的な変動を示します。
責任閾値がどのように機能するかの例として、口唇口蓋裂に見ることができます。口唇口蓋裂は、口唇と口蓋の組織が融合していない状態で生まれてくる先天異常です。口唇口蓋裂とその罹患児は、障害の家族歴を持っているように見えない影響を受けていない両親を持っています。患児の両親が障害を持っていないという事実にもかかわらず、両親は口唇と口蓋形成に必要ではあるが活性は低い遺伝子を持っている可能性があります。実際、口唇口蓋裂の発生率は、罹患した子供のいる家庭で高いので、この先天異常には遺伝的な要素があると考えられています。さらに、いくつかの栄養不足と母親の喫煙は、この出生時の先天異常と関連しているので、環境要因も関与しています。個人が口唇口蓋裂を持って生まれてきた場合、この状態の原因となる要因は、責任の閾値を超えていることになります。閾値をかなり超えると、先天異常は重症度を増します。このような場合には、他の家族も罹患している可能性が高くなります。口唇口蓋裂は、このように不連続的な変動を伴う多因子疾患です。
多因子遺伝のたゆまぬ研究
ヒトでは、他にも多発性硬化症、糖尿病、喘息、癌、多数の先天性欠損症など、多因子遺伝パターンを示す疾患が多数存在します。こうした疾患はいずれも、コピー数変動、エピスタティック相互作用(エピスタティス:遺伝学において、異なる遺伝子座間の相互作用が一つの形質に影響すること)、モディファイア効果などの遺伝的要因と、様々な環境要因との複雑な相互作用に起因しています。不連続な形質変異を持つケースでは、このような多数の要因が責任の閾値を超えることもあれば超えないこともあり、病気が発生するかどうかを予測することは困難です。
これまで、複雑な疾患を研究するために、数多くの方法が開発されてきました。有望な方法の一つは、主要な複合障害の根底にある共通の遺伝的因子を同定するゲノムワイド関連研究(GWAS)の利用です。
今日、それが可能となったにもかかわらず、様々な多因子性疾患の原因と性質については、まだ多くのことが解明されていないのが現状です。
2遺伝子遺伝 digenic inheritance
常染色体劣性遺伝は同じ遺伝子の対立遺伝子が両方とも病的遺伝子になることでおこりますが、これと同じ状況が全く別の遺伝子同士で引き起こされることを2遺伝子遺伝といいます。
20 世紀初頭から遺伝学者を魅了してきたのがこの2遺伝子遺伝(DI)です。遺伝学研究の初期の数十年間では、「エピスタシス(epistatis)」(異なる遺伝子座間の相互作用が一つの形質に影響すること)という用語が一部の遺伝性遺伝のいくつかの形態を説明するために使われていました が、ここ数十年で「エピスタシス(epistasis)」は、ゲノムワイドな関連性研究によって同定された遺伝子座の相互作用を含むが、それに限定されない多遺伝子疾患におけるより広い範囲の遺伝子座-遺伝子座相互作用を説明するために使われるようになりました 。
Defris-Gussenhovenは50年以上前に、遺伝学的解析で単原性として扱われた場合には、浸透性の低下を示すヒトの病気の血統が多く存在するが、その遺伝は 2 遺伝座モデルによってより正確に説明できることを示唆した。ここでいう「浸透率の低下」とは、罹患した血縁者のすべて、あるいはほとんどすべての血縁者が、一次変異遺伝子型を持つとモデル化されているにもかかわらず、一次変異遺伝子型を持つ一人以上の親族は影響を受けていないことを意味しています。
ヒトの疾患における二遺伝子遺伝DIの最初の報告は、1994 年の網膜色素変性症(RP)でした 。1994 年以降、2001 年には、Bardet-Biedl 症候群(BBS)、難聴、その他の表現型におけるヒトにおける二遺伝子遺伝DIの報告が目立つようになりました。
近年では以下に示すように67の疾患で2遺伝子遺伝が報告されています。
| 2遺伝子遺伝の疾患 | 遺伝子/遺伝子座1 | 遺伝子/遺伝子座2 | ||
|---|---|---|---|---|
| QT延長症候群 | KCNH2/7q | KCNQ1/11p | ||
| QT延長症候群 | various LQT genes | various LQT genes | ||
| 難聴 | GJB2/13q | GJB6/13q | ||
| 難聴 | TECTA/11q | 1p | ||
| ペンドレッド症候群(難聴) | SLC26A4/7q | FOXI1/5q | ||
| 難聴 | 1q | 4q | ||
| 難聴 | CDH23/10q | ATP2B2/3p | ||
| アッシャー症候群 | MYO7A/11q | 3q | ||
| アッシャー症候群 | CDH23/10q | PCDH15/10q | ||
| アッシャー症候群 | PDZD7/10q | GPR98/5q | ||
| バーター症候群 (先天性難聴) | CLCNKA/1p | CLCNKB/1p | ||
| バルデ・ビーデル症候群 | BBS2/16q | various BBS loci | ||
| バルデ・ビーデル症候群 | BBS4/15q | various BBS genes | ||
| バルデ・ビーデル症候群 | BBS1/11q | various BBS genes | ||
| ジュベール症候群 (+線毛不全症) | CEP41/7q | various genes | ||
| レーバー先天性黒内障(+線毛不全症) | CEP290/12q | MKKS/BBS6/20p | ||
| 短肋骨多指症候群 (線毛不全症) | NEK1/4q | DYNC2H1/11q | ||
| ネフローゼ症候群 | NPHS1/19q | NPHS2/1q | ||
| 低ゴナドトロピン性性機能低下症 | PROKR2/20p | KAL1/Xp | ||
| 低ゴナドトロピン性性機能低下症 | FGFR1/8p | NSMF/9q | ||
| 低ゴナドトロピン性性機能低下症 | FGF8-related | FGF8-related | ||
| 低ゴナドトロピン性性機能低下症候群 | RNF216/7p | OTUD4/4q | ||
| ヒルシュスプルング病 | RET/10q | EDNRB/13q | ||
| パーキンソン病 | PARK7/1p | PINK1/1p | ||
| 網膜色素変性症 | PRPH2/6p | ROM1/11q | ||
| 若年性緑内障 | MYOC/1q | CYP1B1/2p | ||
| ワールデンブルグ症候群/白皮症 | MITF/3p | TYR/11q | ||
| 眼皮膚白皮症 (OCA) | TYR/11q | OCA2/15q | ||
| 接合部型表皮水疱症 | COL17A1/10q | LAMB3/1q | ||
| 線維素原異常血症 (凝結遅延) | FGA/4q | FGG/4q | ||
| 多発性嚢胞腎 | PKD1/16p | PKD2/4q | ||
| 全前脳胞症 | SHH/7q | TGIF1/18p | ||
| 家族性高コレステロール血症 | LDLR/19p | 13q | ||
| シスチン尿症 | SLC3A1/2p | SLC7A9/19q | ||
| 高インスリン血症 | PPARG/3p | PPP1R3A/7q | ||
| 高コラン酸血症 | TJP2/9q | BAAT/9q | ||
| 褐色細胞腫 | TMEM127/2q | 16p | ||
| 家族性滲出性硝子体網膜症 | FZD4/11q | F5/1q | ||
| 第Ⅷ因子性血栓 | 5q | 11q | ||
| PMP22関連神経症 | PMP22/17p | various genes | ||
| シャルコ・マリー・ツース病 (非PMP22) | MFN2/1p | GDAP1/8q | ||
| エメリー・ドレイフス型筋ジストロフィー | LMNA/1q | EMD/Xq | ||
| ポルフィリン症 (急性) | various genes | HFE/6p | ||
| ポルフィリン症 (急性) | UROD/1p | HMBS/11q | ||
| ポルフィリン症 (急性) | CPOX/3q | ALAD/9q | ||
| て ん か ん熱 性 け い れ ん プ ラ ス | 1q | 18q | ||
| ヘモクロマトーシス | HFE/6p | HAMP/19q | ||
| 進行性外眼筋麻痺 | C10orf2/10q | POLG/15q | ||
| て ん か ん熱 性 け い れ ん プ ラ ス | SCN1A/2q | SCN2A/2q | ||
| 光感受性てんかん | 7q | 16p | ||
| 裂手/裂足症 | 1q | 6q | ||
| イミノグリシン尿症 | SLC36A2/5q | SLC6A20/3p | ||
| 円錐角膜 | 1p | 8q | ||
| 肢帯型筋ジストロフィー | SGCB/4q | SGCD/5q | ||
| ウルリヒ型先天性筋ジストロフィー | COL6A1/21q | COL6A2/21q | ||
| 弾性線維性仮性黄色腫 | ABCC6/16p | GGCX/2p | ||
| 遺伝性運動神経症 | DSCL2/11q | 16p | ||
| 口唇裂 | 1q | 2p | ||
| フックス角膜内皮ジストロフィー | ZEB1/10p | 9p | ||
| 遺伝性前眼部形成異常 | PITX2/4q | FOXC1/6p | ||
| 大腸がん | MUTYH/1p | OGG1/3p | ||
| ローター症候群 (高ビリルビン血症) | SLCO1B1/12p | SLCO1B3/12p | ||
| デント病 | CLCN5/Xp | OCRL/Xq | ||
| 顔面肩甲上腕型筋ジストロフィー | DUX4/4q | SMCHD1/18p | ||
| 単純型表皮水疱症 | KRT5/12q | KRT14/17q | ||
| 悪性黒色腫易罹患性 | CDKN2A/9p | MC1R/16q | ||
| 寡毛症 (非症候性) | CDH3/16q | 12q |







