目次

📍 クイックナビゲーション
ABCDトランスポーターとは、私たちの細胞の中にあるペルオキシソームやリソソームという小さな器官に特化した4種類の膜タンパク質(ABCD1・ABCD2・ABCD3・ABCD4)の総称です。ATPのエネルギーを使って極長鎖脂肪酸やビタミンB12といった特殊な物質を細胞内で輸送し、この遺伝子に変異が生じると副腎白質ジストロフィーなどの深刻な遺伝性疾患を引き起こします。2026年現在、遺伝子治療や新規薬剤など、治療開発が歴史的な転換点を迎えています。
Q. ABCDトランスポーターとはどういうタンパク質ですか?まず結論だけ知りたいです
A. ペルオキシソームとリソソームに局在し、ATPのエネルギーで極長鎖脂肪酸・分岐鎖脂肪酸・ビタミンB12などを細胞内輸送するABC輸送タンパク質スーパーファミリーのサブグループ(ABCD1〜4)です。これらの遺伝子に変異が生じると、X連鎖副腎白質ジストロフィー・先天性胆汁酸合成異常症5型・cblJ型メチルマロン酸血症といった進行性の代謝・神経変性疾患を引き起こします。
- ➤タンパク質の定義と進化 → 全生物に保存された巨大ファミリーの中で細胞内オルガネラに特化したサブグループ
- ➤原子レベルの分子構造 → Cryo-EMで解明された前例のない輸送サイクルと基質認識機構
- ➤ABCD1〜4それぞれの機能 → 局在・基質・相互作用因子の違いと代償メカニズム
- ➤関連遺伝性疾患 → X-ALD・CBAS5・cblJ型の分子基盤と病態
- ➤2026年現在の最新治療 → 遺伝子治療・PPAR-γ作動薬・TR-β作動薬の3つの革新的アプローチ
1. ABCDトランスポーターとは:生命に普遍的なABCスーパーファミリーの一員
ABCトランスポータースーパーファミリーは、細菌・古細菌・真核生物のすべてにわたって存在する、地球上で最も広く保存された膜タンパク質群のひとつです。ヒトのゲノムには48種類のABCトランスポーター遺伝子が含まれており、遺伝子構造やアミノ酸配列の相同性に基づいてABCAからABCIまでのサブファミリーに分類されています。その中で今回取り上げるABCDサブファミリー(ABCD1〜4)は、主にペルオキシソームとリソソームという細胞内小器官に特化した役割を担うよう進化してきた、特別なグループです。
💡 用語解説:ABCトランスポーターとは
ABC(ATP-Binding Cassette:ATP結合カセット)トランスポーターとは、ATPという細胞のエネルギー通貨を使って、細胞膜や細胞内小器官の膜を越えてさまざまな物質を能動的に運ぶタンパク質の総称です。すべてのABCトランスポーターは共通して2つの膜貫通ドメイン(TMD)と2つのヌクレオチド結合ドメイン(NBD)という4つのコアドメインを持ちます。TMDが物質の通り道を作り、NBDがATPを結合・分解してエネルギーを取り出す役割を担います。大腸菌のゲノムには約80種、出芽酵母には31種、そしてヒトには48種のABCトランスポーター遺伝子が含まれています。
ABCトランスポーターが輸送する物質は極めて多様で、栄養素・脂質・胆汁酸・ビタミン・薬物・毒素・ペプチドなど多岐にわたります。私たちがよく聞く「多剤耐性」(抗がん剤が効かなくなる現象)の一因も、ABCトランスポーターが薬物を細胞の外へ排出してしまうことにあります。一方、ABCDサブファミリーは薬物耐性とは無関係で、細胞内オルガネラ間での脂質や補酵素の輸送という精密な生理機能に特化しています。
💡 用語解説:ペルオキシソームとリソソームとは
ペルオキシソームは細胞内に存在する小さな膜に包まれた器官で、極長鎖脂肪酸(炭素数22以上の長い脂肪酸)や分岐鎖脂肪酸をβ酸化という反応で分解したり、胆汁酸の生合成に関わる脂質中間体を処理したりする「代謝の工場」です。ペルオキシソームの構造・機能・関連疾患全般についてはペルオキシソームの全貌をご覧ください。
リソソームは細胞内の「消化・リサイクルセンター」で、エンドサイトーシス(細胞が外から物質を取り込む仕組み)によって取り込まれたタンパク質や複合体を分解し、その構成成分を細胞質へ放出する役割を担います。ABCDサブファミリーはこれら2つのオルガネラに局在し、それぞれで異なる基質を輸送します。
2. 原子レベルで解明された分子構造と輸送メカニズム
近年のクライオ電子顕微鏡(Cryo-EM)技術の飛躍的進歩により、ABCDトランスポーターが巨大かつ疎水性の高い基質をどのように認識・輸送するかという分子メカニズムが、原子レベルで明らかにされつつあります。
💡 用語解説:クライオ電子顕微鏡(Cryo-EM)とは
タンパク質を超低温(マイナス180℃以下)で急速凍結し、電子線を当てて撮影した大量の2次元画像をコンピューターで解析することで、タンパク質の3次元構造をオングストローム(0.1ナノメートル)レベルの解像度で決定する技術です。2017年のノーベル化学賞を受賞した革命的な手法で、X線結晶構造解析では困難だった膜タンパク質の構造解析を可能にしました。
ABCD1の独特な輸送メカニズム:アシル鎖を脂質膜に「逃がす」戦略
ABCD1(別名ALDP:Adrenoleukodystrophy Protein)は、極長鎖脂肪酸(VLCFA)のCoAチオエステルを細胞質からペルオキシソーム内腔へ輸送します。リン脂質ナノディスクに再構成されたヒトABCD1の高解像度Cryo-EM解析(最大3.35Å)により、複数の立体構造(アポ状態・基質結合状態・ATP結合状態)が明らかになりました。
💡 用語解説:極長鎖脂肪酸(VLCFA)・CoAチオエステルとは
極長鎖脂肪酸(VLCFA:Very Long-Chain Fatty Acid)とは、炭素数22以上の非常に長い鎖を持つ脂肪酸のことです。C24:0(リグノセリン酸)やC26:0(セロチン酸)が代表例で、ミエリン(神経の絶縁体)や細胞膜の構成成分として重要ですが、蓄積すると神経毒性を持ちます。
CoAチオエステルとは、脂肪酸が補酵素A(CoA)と結合した活性化された形です。細胞内での脂肪酸代謝はCoAチオエステルの形で進行します。ABCD1が輸送するのはVLCFA単体ではなく、このVLCFA-CoAという活性化された形です。
構造解析で判明した最も重要な発見は、ABCD1が他のABCトランスポーターとは全く異なる輸送の「戦略」をとることです。2つの対称的なC22:0-CoA分子が膜貫通ドメイン(TMD)に協同的に結合する際、親水性である3′-ホスホ-ADP部分のみが膜貫通キャビティ(空洞)に入り込む一方、非常に疎水性の高い長大な脂肪酸アシル鎖は周囲の脂質二重層の環境へと「逃がされる」という前例のないメカニズムです。さらに、第5膜貫通ヘリックスに位置するトリプトファン残基(W339)が基質の結合とATP加水分解の刺激に必須であること、またABCD1特有のC末端コイルドコイルドメインがATPase活性を負に調節することも明らかになっています。
ABCD3の構造とパラダイムシフト:「インポーター」としてのABCD4
ABCD3についても、アポ状態(3.33Å)とフィタノイルCoA結合状態(3.13Å)のCryo-EM構造が解明されており、基質結合がATPase活性を強く誘導することが確認されています。そして最も革命的な発見のひとつが、ABCD4がリソソーム内腔から細胞質へとコバラミン(ビタミンB12)を輸送する「インポーター」として機能することです。
💡 用語解説:インポーターとエクスポーターのパラダイムシフト
長らく「真核生物のABCトランスポーターはすべてエクスポーター(物質を排出するポンプ)として機能する」という定説がありました。ところがABCDサブファミリーの詳細な解析により、この定説が修正を迫られています。ABCD1〜3はペルオキシソームへの「取り込み(インポート)」に関与し、特にABCD4はリソソームから細胞質への方向で機能する純粋な「インポーター」であることが証明されました。真核生物のABCトランスポーターでインポーターが確認されたことは、膜輸送生物学における大きなパラダイムシフトです。
3. ABCD1〜4それぞれの機能と細胞内局在
ABCD1・ABCD2・ABCD3の3つは主にホモ二量体としてペルオキシソーム膜に局在しています。これらは遊離ポリソームで翻訳された後、N末端付近の「H0モチーフ」をシャペロンタンパク質Pex19pに認識され、ペルオキシソーム膜へと組み込まれます。一方ABCD4はH0モチーフを持たず、小胞体経由でリソソームへ輸送されるという、全く異なる経路をたどります。
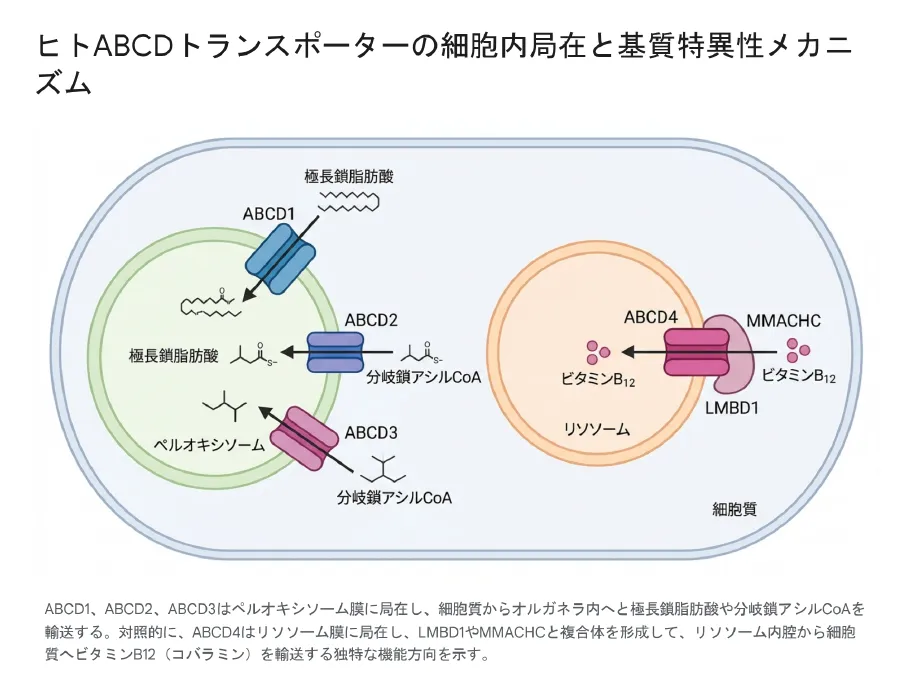
ABCD1・ABCD2・ABCD3はペルオキシソーム膜に局在し細胞質からオルガネラ内へ極長鎖脂肪酸や分岐鎖アシルCoAを輸送する。対照的にABCD4はリソソーム膜に局在し、LMBD1やMMACHCと複合体を形成してリソソーム内腔から細胞質へビタミンB12(コバラミン)を輸送する独特の機能方向を示す。
🔵 ABCD1(ALDP)
- 局在:ペルオキシソーム膜
- 主な基質:飽和・一価不飽和VLCFA-CoA(特にC24:0、C26:0)
- 標的化:H0モチーフ → Pex19p
- 関連疾患:X連鎖副腎白質ジストロフィー(X-ALD)
🟣 ABCD2
- 局在:ペルオキシソーム膜
- 主な基質:多価不飽和VLCFA-CoA(特にC22:6、C24:6)
- ABCD1との同一性:アミノ酸配列約66%
- 特記:ABCD1欠損時に代償的に機能しうる
🟢 ABCD3(PMP70)
- 局在:ペルオキシソーム膜
- 主な基質:分岐鎖アシルCoA、LCFA-CoA、胆汁酸中間体(THCA・DHCA)
- 関連疾患:先天性胆汁酸合成異常症5型(CBAS5)
🟡 ABCD4
- 局在:リソソーム膜(小胞体経由で輸送)
- 主な基質:コバラミン(ビタミンB12)
- 相互作用:LMBD1・MMACHC複合体
- 関連疾患:cblJ型メチルマロン酸血症・ホモシスチン尿症
ABCD1とABCD2の基質のオーバーラップと「代償作用」
ABCD1とABCD2は輸送する基質の一部が重複しています。ABCD1はC24:0-CoAやC26:0-CoAに対して高い特異性を示す一方、ABCD2はドコサヘキサエン酸(DHA)などの多価不飽和脂肪酸(C22:6-CoA・C24:6-CoA)に対してより高い親和性を持ちます。両者は約66%のアミノ酸配列同一性を持ち、ABCD1が欠損した病的状態でABCD2を過剰発現させると、VLCFAのペルオキシソームへの輸送が代償的に回復することが、細胞モデルおよびトランスジェニックマウスモデルで証明されています。この「代謝的代償作用」が、X-ALDの治療戦略のひとつの科学的根拠となっています(詳しくは第6章)。
ABCD4の精巧なコバラミン輸送複合体
ABCD4によるコバラミン輸送は、単体のタンパク質ではなく、精緻なタンパク質複合体によって駆動されます。リソソーム膜においてABCD4はLMBD1と1:1の化学量論比で特異的に結合し複合体を形成します。LMBD1はABCD4のリソソームへの輸送を担うシャペロンであり、コバラミン輸送の触媒活性はABCD4自身が持っています。
さらにこの複合体は、細胞質のコバラミン結合タンパク質であるMMACHCと低ナノモルオーダーの高い親和性(KD:LMBD1が約14 nM、ABCD4が約65 nM)で相互作用します。このMMACHCへの直接的な引き渡し(ベクトル輸送)により、リソソームから放出されたコバラミンが細胞質に拡散・希釈されることなく確実にMMACHCへ届けられます。最終的にコバラミンはアデノシルコバラミン(AdoCbl)とメチルコバラミン(MeCbl)へと変換され、それぞれメチルマロニルCoAムターゼとメチオニンシンターゼの補酵素として機能します。
💡 用語解説:コバラミン(ビタミンB12)とその役割
コバラミン(ビタミンB12)は、コバルトを含む水溶性ビタミンで、体内で2種類の活性型補酵素(AdoCblとMeCbl)に変換されます。AdoCblはメチルマロニルCoAムターゼの補酵素としてアミノ酸・脂肪酸・コレステロールの分解に関与し、MeCblはメチオニンシンターゼの補酵素としてホモシステインからメチオニンへの変換(葉酸代謝サイクル)に不可欠です。これらが欠乏すると、メチルマロン酸やホモシステインが蓄積し、重篤な代謝障害を引き起こします。

4. ABCDトランスポーター変異が引き起こす遺伝性疾患
ABCDサブファミリーの機能不全は、進行性の代謝性・神経変性疾患の直接的な原因となります。各メンバーの遺伝子変異が引き起こす疾患を解説します。
① X連鎖副腎白質ジストロフィー(X-ALD):最も頻度の高いペルオキシソーム異常症
X染色体上に位置するABCD1遺伝子の変異は、出生数約17,000人に1人(男女含む)の頻度で見られる最も一般的なペルオキシソーム異常症「X連鎖副腎白質ジストロフィー(X-ALD:OMIM 300100)」を引き起こします。ABCD1タンパク質の機能不全により、VLCFAがペルオキシソームで分解されず、血漿・脳のグリア細胞・脊髄・副腎などに毒性レベルで蓄積します。
💡 用語解説:X連鎖遺伝とは
X連鎖(X染色体連鎖)遺伝とは、原因遺伝子がX染色体上にある遺伝形式です。女性(XX)は2本のX染色体を持つため、片方のX染色体に変異があっても、もう片方の正常な遺伝子で補われ、無症状または軽症のキャリア(保因者)になることが多いです。しかし男性(XY)はX染色体が1本しかないため、変異遺伝子を受け継ぐと補完できず重篤な症状が出ます。X-ALDが主に男性に重症発症する理由はこれです。なお女性キャリアでも成人後に症状(AMNに似た神経症状など)が現れることがあります。
X-ALDの表現型は極めて多様で、同じABCD1変異でも全く異なる経過をたどります。特に注目すべき2つの主要な表現型があります。
🚨 小児大脳型ALD(CCALD)約30%
- 発症年齢:4〜8歳
- 中枢神経の激しい炎症性脱髄
- 発症から半年〜2年で植物状態または死亡
- 最も過酷な病型
⚠️ 副腎脊髄ニューロパチー(AMN)約40%
- 発症年齢:成人(20代以降)
- 脊髄の非炎症性軸索変性
- 5〜15年かけて歩行障害が進行
- 最終的に車椅子が必要になることも
② 先天性胆汁酸合成異常症5型(CBAS5)
ABCD3遺伝子の常染色体劣性変異は、肝臓が正常な胆汁酸を生成できず胆汁うっ滞を引き起こす稀な疾患「先天性胆汁酸合成異常症5型(CBAS5:OMIM 616278)」の原因です。ABCD3欠損により胆汁酸前駆体であるTHCAやDHCAのペルオキシソームへの移行が阻害され、正常な胆汁酸生合成経路が完全に破綻します。臨床的には黄疸・肝腫大・脾腫・肝線維化・鉄欠乏性貧血などの多彩な症状が見られます。
💡 用語解説:常染色体劣性遺伝とは
常染色体劣性遺伝(AR:Autosomal Recessive)とは、両親からそれぞれ1つずつ変異遺伝子を受け継いだ場合(ホモ接合または複合ヘテロ接合の状態)にのみ発症する遺伝形式です。両親は通常それぞれ1つの変異遺伝子を持つ「キャリア(保因者)」で、自身は無症状です。子どもへの遺伝確率は25%(発症)・50%(キャリア)・25%(正常)となります。CBAS5やcblJ型はこのAR遺伝形式をとります。
③ cblJ型メチルマロン酸血症・ホモシスチン尿症
ABCD4遺伝子の常染色体劣性変異は、「cblJ型メチルマロン酸血症・ホモシスチン尿症」を引き起こします。機能的なABCD4タンパク質が欠損するとLMBD1-MMACHC複合体によるリソソームからのビタミンB12の放出が遮断され、細胞質にAdoCblとMeCblの深刻な欠乏が生じます。その結果、重度の発達遅滞・眼科的欠損・神経学的問題・巨赤芽球性貧血・皮膚の色素沈着異常などが引き起こされます。
コバラミン代謝異常症はcblJ型以外にも複数の種類があり、原因遺伝子によって分類されます。以下に関連疾患と遺伝子ページへのリンクをまとめています。
cblJ型(ABCD4)・cblC型(MMACHC)・cblD型・cblF型・cblB型(MMAB)・cblX型(HCFC1・THAP11)・cblC型ジェニック
遺伝子ページ:MMAB・HCFC1・THAP11
ペルオキシソームβ酸化障害とミトコンドリアβ酸化障害の違い
脂肪酸のβ酸化はペルオキシソームとミトコンドリアの両方で行われますが、担う脂肪酸の「鎖長」が異なります。ABCDトランスポーター関連疾患はペルオキシソームでの極長鎖脂肪酸(炭素数22以上)の処理障害です。一方、ミトコンドリアで中鎖脂肪酸のβ酸化を担うACADM(中鎖アシルCoA脱水素酵素)遺伝子の変異による中鎖アシルCoA脱水素酵素欠損症(MCAD欠損症)は、蓄積する基質・主な症状・治療アプローチが全く異なる別疾患です。両者を混同しないよう、代謝経路を理解したうえでの鑑別が重要です。
④ 重要な鑑別:「ABCD症候群」はABCDトランスポーターとは無関係
名称の類似性から混同されることがありますが、「ABCD症候群」はABCトランスポーターサブファミリーDとは全く無関係の別疾患です。ABCD症候群は白皮症(Albinism)・黒毛束(Black lock)・腸管神経細胞遊走異常(Cell migration disorder of neurocytes of the gut:ヒルシュスプルング病)・難聴(Deafness)の頭文字をとった、神経堤細胞由来組織の発生障害に起因する超希少疾患です。発症機序はABCDトランスポーターの膜輸送障害とは根本的に異なります。
5. X連鎖副腎白質ジストロフィーの病態生理:動物モデルが示した新知見
X-ALD(副腎白質ジストロフィー)の最大の臨床的謎は、同一のABCD1変異を持つ患者の間でも(同じ家系の兄弟間でも)、表現型が著しく異なることです。小児大脳型で急速に進行する場合もあれば、成人になるまで症状が出ない場合もあります。この謎の解明において、動物モデルの研究が大きな役割を果たしています。
なお白質ジストロフィーにはX-ALD以外にも複数の原因疾患があります。脂質蓄積型として異染性白質ジストロフィー(MLD)、低髄鞘化型としてペリツェウス・メルツバッハ病(PMD)などが知られています。X-ALDは脱髄(ミエリンの炎症性破壊)を特徴とする点で、これらの低髄鞘化疾患とは病態メカニズムが異なります。
マウスモデルでは、Abcd1単独ノックアウトマウスはヒトのAMN(成人発症の脊髄疾患)に似た遅発性の脊髄軸索変性を示しますが、小児大脳型のような激しい脳内炎症は自然発症しません。重要な知見は、このマウスの脊髄オリゴデンドロサイトにABCD1遺伝子を導入すると、遅発性の軸索病変が急速に可逆的になるという事実です。これはミエリンを形成するオリゴデンドロサイトが軸索へのエネルギー基質供給に極めて重要であることを示しています。
💡 用語解説:疾患修飾因子(Modifier genes)とは
主たる原因遺伝子の変異以外に、疾患の重症度や表現型に影響を与える他の遺伝子や環境因子のことです。X-ALDでは ABCD1変異が「原因遺伝子」ですが、なぜ同じ変異でも子どもに重症の脳型が出る場合と成人になるまで症状が出ない場合があるのか、その理由は未解明です。ホモログであるABCD2・ABCD3・ABCD4が修飾因子になりうる可能性も研究されていますが、現時点では主要な修飾遺伝子であるとは言い切れず、解明は次世代シーケンシング等を用いた継続的な研究課題となっています。また、外傷・感染などの環境的トリガーや確率論的なグリア細胞間クロストークの差異も病態決定に関わると考えられています。
Abcd1とAbcd2の二重欠損(ダブルノックアウト)マウスの腹腔マクロファージを用いた研究では、Abcd2単独欠損ではVLCFAのβ酸化はほとんど損なわれませんが、二重欠損ではVLCFAの蓄積が劇的に悪化することが明らかになりました。ヒトのX-ALD単球はABCD2の発現を欠いているため重篤な代謝表現型を示しますが、マウスのマクロファージでは内在性ABCD2発現が病態の深刻化を防いでいると考えられます。
ショウジョウバエのノックアウトモデルや、ペルオキシン-5(peroxin-5)KOマウスモデル(オリゴデンドロサイト特異的にペルオキシソーム生合成を阻害)では、ヒトの大脳型ALDのほぼ完璧な表現型模写が達成されます。これらの知見から、X-ALDの病態においてオリゴデンドロサイトと軸索の病理が「主役」であり、ミクログリアの活性化による神経炎症が「二次的な増悪因子」として働くという新たなパラダイムが提示されています。なお、VLCFAの蓄積に伴う酸化ストレスについては、ペルオキシソームのアンチオキシダント系(PRDX1などのペルオキシレドキシン)との関連も研究されています。
6. X-ALDに対する次世代治療開発(2026年現在)
X-ALD、特に小児大脳型(CCALD)は進行が極めて速く致死的な疾患です。現在確立された治療法は、神経症状が軽微な早期段階で実施される造血幹細胞移植(HSCT)に限られています。しかし2026年現在、全く異なる薬理学的作用機序を持つ3つの革新的なアプローチが臨床開発の最終段階でしのぎを削っています。
アプローチ①:遺伝子治療(SKYSONA™・レンチウイルスIT-IV・SBT101)
機能的なABCD1遺伝子を患者の細胞へ直接導入する治療法です。
SKYSONA™(早期CCALD男児対象)
患者自身の造血幹細胞をレンチウイルスベクターで体外改変し戻す自己ex vivo遺伝子治療。早期投与で神経後遺症を最小化できる可能性。ただし治療患者の約10%において挿入変異に起因する血液悪性腫瘍(骨髄異形成症候群・急性骨髄性白血病)の報告あり。長期安全性の監視が必須。
レンチウイルスIT-IV(NCT03727555)
正常ABCD1遺伝子を搭載した自己不活化型レンチウイルスベクターを髄腔内(IT)または静脈内(IV)に直接注射するin vivo遺伝子治療。2025年後半〜2028年にかけてデータ収集が予定されている進行中の試験。FDAに承認されたin vivoレンチウイルス遺伝子治療は未だ存在しない。
SBT101(AAV9ベクター)
成人AMN患者を対象としたAAV9を用いたin vivo遺伝子治療(Spur Therapeutics社)。ゲノム統合を行わず発がんリスクが低い理論的利点があったが、この特定の臨床試験は2025年10月末日をもって終了(terminated)。
アプローチ②:レリグリタゾン(NEZGLYAL®):NEXUS試験の成功とEMAへの再申請
Minoryx Therapeutics社が開発したレリグリタゾン(商品名:NEZGLYAL®)は、血液脳関門を効率的に通過する選択的PPAR-γ(ペルオキシソーム増殖因子活性化受容体γ)作動薬です。
💡 用語解説:PPAR-γ(ペルオキシソーム増殖因子活性化受容体γ)とは
PPAR-γは細胞核内にある転写因子(タンパク質の設計図を読む調節役)で、脂肪細胞の分化・糖代謝・炎症抑制に関わります。この受容体を活性化する薬(作動薬=アゴニスト)は糖尿病治療薬(チアゾリジン系薬)としても知られていますが、レリグリタゾンは血液脳関門を通過できるよう設計されており、脳内でのPPAR-γ活性化を通じた神経炎症抑制・ミトコンドリア機能改善・酸化ストレス軽減を狙いとしています。
欧州医薬品庁(EMA)はADVANCE試験の結果をもとに2024年1月に「承認拒否」勧告を出し、2024年5月に最終的に拒否が確定しました。主な理由は6分間歩行テストでの有意な改善が示せなかったことなどです。しかし事態はその後劇的に好転します。
🌟 NEXUS試験(小児cALD、96週間)の結果(2024年12月発表)
- 対象:小児の初期cALD男児23名(評価対象20名)
- 主要評価項目を達成:評価対象20名全員が治療中に臨床的に安定
- 7名(35%)が疾患停止(arrested disease)基準を満たした(自然停止率10%を有意に上回る:p<0.05)
- 全患者でレリグリタゾンの忍容性は良好。治療関連重篤有害事象ゼロ
- この結果を受け、2025年にEMAへ新たな販売承認申請(MAA)を提出
- 2025年7月、EMAがMAAを正式に受理(Validation)し審査進行中
アプローチ③:VK0214:TR-β作動薬によるABCD2代償的発現誘導療法
Viking Therapeutics社が開発中のVK0214は、甲状腺ホルモン受容体β(TR-β)に選択的に結合する経口低分子化合物です。ABCD2遺伝子プロモーター領域には機能的な甲状腺ホルモン応答配列(TRE)があり、TR-βを活性化することでABCD2の転写を強力に促進します。欠損したABCD1の代わりにABCD2にVLCFAのペルオキシソーム輸送を代償させるという戦略です。
💡 用語解説:TR-β(甲状腺ホルモン受容体β)選択的作動薬とは
天然の甲状腺ホルモン(T3)はTR-αとTR-βの両方を活性化しますが、TR-αを活性化すると心毒性(頻脈・心不全)などの副作用が生じます。VK0214はTR-βにのみ選択的に結合するよう設計されており、ABCD2遺伝子の発現誘導という目的(TR-β依存的)を達成しながら、TR-α関連の心毒性副作用を回避できます。
AMN成人男性患者を対象とした第1b相臨床試験では、VK0214の28日間1日1回経口投与が良好な安全性・忍容性プロファイルを示し、プラセボ群と比較して血漿中VLCFAレベルが統計学的に有意に減少するという臨床的概念実証(POC)が達成されました。in vivoモデルでは脳で35%(p<0.05)、肝臓で262%(p<0.05)のABCD2 mRNA発現増加が確認されており、長期的な神経変性抑制効果を検証するためのさらなる臨床研究が進行中です。
7. 遺伝カウンセリングの重要性
ABCDトランスポーター関連疾患の確定診断後は、家族への丁寧な遺伝カウンセリングが不可欠です。
X-ALDにおける遺伝カウンセリングの特殊性
X-ALDはX連鎖遺伝のため、女性キャリア(保因者)の認識が特に重要です。ABCD1変異を持つ母親は、息子に50%の確率でX-ALDを伝える可能性があります。また、女性キャリア自身も成人後にAMNに類似した神経症状や副腎症状が現れることがあります。
- ➤保因者(キャリア)スクリーニング:家族歴がある女性や、次子の妊娠を考えている方にはキャリアスクリーニングが推奨されます。X-ALDに関しては米国人類遺伝学会(ACMG)・産婦人科学会(ACOG)も保因者スクリーニングを推奨しており、その重要性は国際的に認められています。
- ➤表現型の多様性について:同じ変異でも病型が予測できない点を丁寧に説明し、新生児スクリーニング(一部地域で実施中)や定期的な副腎機能・MRI評価の重要性を伝えます。
- ➤家族計画について:ALDと診断された場合でも、着床前遺伝学的検査(PGT)や出生前診断などの選択肢があります。診断後すぐに家族計画を諦める必要はありません。
AR疾患(CBAS5・cblJ型)における遺伝カウンセリング
常染色体劣性疾患では、両親ともにキャリアであることが多く、次子への遺伝確率は25%です。遺伝子検査による確認と出生前診断の選択肢について、臨床遺伝専門医が詳しく説明します。核・ミトコンドリアNGS遺伝子検査により、家系内変異の確認や保因者診断が可能です。
8. よくある誤解
誤解①「ABCD症候群=ABCDトランスポーター疾患」
全く異なる疾患です。ABCD症候群は神経堤細胞の発生障害(白皮症・難聴・ヒルシュスプルング病など)であり、ABCDトランスポーター遺伝子変異による脂質代謝・コバラミン輸送障害とは発症機序が根本的に異なります。
誤解②「X-ALDは男性だけの病気」
女性キャリアも成人後にAMNに類似した神経症状(歩行障害・膀胱機能障害など)や副腎症状が現れることがあります。「女性は症状が出ない」は誤りで、定期的な経過観察が女性キャリアにも推奨されます。
誤解③「cblJ型はビタミンB12を摂れば治る」
cblJ型はビタミンB12の摂取量の問題ではなく、リソソームから細胞質へのコバラミン輸送システムそのものの欠損です。食事でいくらB12を摂取しても、ABCD4の機能なしにはビタミンが活用されません。経路全体の理解が重要です。
誤解④「同じ遺伝子変異なら同じ症状が出る」
X-ALDは全くの逆で、同じABCD1変異を持つ兄弟でも一方が小児大脳型、他方がAMN型になりうることが多数報告されています。遺伝子型と表現型の相関は確立されておらず、これがX-ALDの最大の臨床的謎のひとつです。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 ABCDトランスポーター関連疾患の診断・遺伝カウンセリングについて
X-ALD・cblJ型・CBAS5をはじめとするABCDトランスポーター関連遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] Schmitt L, Tampé R. Structure and mechanism of ABC transporters. Curr Opin Struct Biol. 2002;12(6):754-760. [全文PDF]
- [2] Guenzel AJ, et al. ABC Transporter Subfamily D: Distinct Differences in Behavior between ABCD1-3 and ABCD4 in Subcellular Localization, Function, and Human Disease. Mol Genet Metab. 2016;117(2):S53. [PMC5059523]
- [3] Cryo-EM structure of human very long-chain fatty acid ABC transporter ABCD1 (PDB: 7VWC). [PDBj 7VWC]
- [4] Molecular mechanism of substrate transport by human peroxisomal ABCD3. Proc Natl Acad Sci USA. 2025;122. [PNAS]
- [5] Kawaguchi K, et al. The lysosomal protein ABCD4 can transport vitamin B12 across liposomal membranes in vitro. J Biol Chem. 2021;296:100649. [PMC8113721]
- [6] Mosser J, et al. The genetic landscape of X-linked adrenoleukodystrophy: inheritance, mutations, modifier genes, and diagnosis. Ther Adv Chronic Dis. 2020. [Dove Medical Press]
- [7] Raas Q, et al. Revisiting the Pathogenesis of X-Linked Adrenoleukodystrophy. Genes (Basel). 2025;16(5):590. [MDPI]
- [8] Morita M, et al. Abcd2 Is a Strong Modifier of the Metabolic Impairments in Peritoneal Macrophages of Abcd1-Deficient Mice. PLoS One. 2014;9(9):e107928. [PMC4177892]
- [9] Minoryx Therapeutics. Leriglitazone has met the primary endpoint in NEXUS, the pivotal trial for pediatric patients with cerebral Adrenoleukodystrophy. December 2024. [Minoryx公式PDF]
- [10] Viking Therapeutics. VK0214 Pipeline Information. [Viking Therapeutics公式]
- [11] OMIM #300100. Adrenoleukodystrophy, X-Linked; ALD. Johns Hopkins University. [OMIM]
- [12] OMIM #616278. Bile Acid Synthesis Defect, Congenital, 5; CBAS5. Johns Hopkins University. [OMIM]
- [13] MedlinePlus Genetics. ABCD4 gene. National Library of Medicine. [MedlinePlus]
- [14] Minoryx’s Leriglitazone MAA for cALD Validated by EMA. United Leukodystrophy Foundation. July 2025. [ULF]






