目次

副腎白質ジストロフィー(Adrenoleukodystrophy: ALD)は、ABCD1遺伝子の異常によって体内に「極長鎖脂肪酸(VLCFA)」が蓄積し、脳・脊髄・副腎を障害する遺伝性疾患です。男の子に発症する小児大脳型(cCALD)は治療なしでは数年で命に関わる状態となる一方、成人型の副腎脊髄ニューロパチー(AMN)は何十年もかけて緩やかに進行します。さらに、「保因者」とされてきた女性の半数以上も実際にはAMNを発症することが2024年のガイドラインでついに正式に認められました。新生児スクリーニングの普及と新規治療薬の登場により、この疾患をめぐる医療は今まさに大きな転換点を迎えています。
Q. 副腎白質ジストロフィーとはどんな病気ですか?まず結論だけ知りたい
A. ABCD1遺伝子の異常により極長鎖脂肪酸(VLCFA)が全身に蓄積し、脳・脊髄・副腎を障害するX染色体連鎖の遺伝性疾患です。同じ遺伝子変異でも病型が全く異なり、小児期に激烈な脳の炎症性脱髄を起こす型と成人期に脊髄が緩やかに変性するAMN型があります。女性保因者の半数以上も発症することが2024年以降の定説となっています。
- ➤疾患の定義 → OMIM 300100 / ABCD1遺伝子(Xq28)変異 / 出生男児10万人に1〜2名
- ➤主な病型 → 小児大脳型(cCALD)・成人発症型(AMN)・女性のAMN・副腎皮質機能低下症
- ➤診断方法 → 血漿VLCFA・C26:0-LPC・ABCD1遺伝子解析・Loesスコア・新生児スクリーニング
- ➤最新治療 → 造血幹細胞移植(HSCT)・遺伝子治療Skysona・新規経口薬レリグリタゾン・多職種対症療法
- ➤2024年最新知見 → 女性保因者の積極的管理の義務化・Skysonaの発がんリスク15%問題・レリグリタゾンEMA申請中
1. 副腎白質ジストロフィー(X-ALD)とは
X連鎖性副腎白質ジストロフィー(X-linked Adrenoleukodystrophy: X-ALD、OMIM 300100)は、遺伝性ペルオキシソーム代謝疾患の中で最も頻度が高い疾患です。中枢神経系における激しい炎症性脱髄、脊髄の緩やかな軸索変性、副腎皮質機能低下症を主な症状とします。出生男児10万人あたり約1〜2名、男女合わせると10万人あたり約6名の有病率と推定されており、決して超希少ではない遺伝性疾患です。
💡 用語解説:X-ALD(副腎白質ジストロフィー)の名前の意味
「副腎」=ストレス対応ホルモンを作る臓器。「白質」=脳内の神経線維が集まる領域(髄鞘で覆われている)。「ジストロフィー」=変性・萎縮を意味する医学用語。つまり「副腎と脳の白質が変性する遺伝病」という意味です。「X-ALD」の「X」はX染色体に原因遺伝子があることを示します。
この疾患のもっとも難解な特徴は、「同じ遺伝子変異を持つ兄弟でも、全く異なる病型を呈する」という点にあります。遺伝子型から将来の病型を予測することが現時点では不可能であり、これが適切な治療介入のタイミング決定を難しくしています。
💡 用語解説:X連鎖劣性遺伝(Xリンク遺伝)とは
原因遺伝子がX染色体上にあります。男性はX染色体を1本しか持たないため、その1本に変異があれば発症します。女性はX染色体を2本持ち、1本に変異があっても多くの場合もう1本でカバーされます(保因者)。ただしX-ALDでは、女性保因者の半数以上が生涯のうちにAMNを発症することが現在では明確にわかっています。「保因者だから安心」という認識は過去のものです。
💡 用語解説:VLCFA(極長鎖脂肪酸)とは
VLCFA(Very Long-Chain Fatty Acids)とは、炭素数が22以上の非常に長い直鎖脂肪酸のことです。代表的なものにヘキサコサン酸(C26:0)があります。通常はペルオキシソームという細胞小器官で分解されますが、X-ALDではその分解ができず、脳の白質・脊髄・副腎・精巣などに異常蓄積し、細胞を傷つけます。血液検査でVLCFAの濃度を測定することがX-ALDの基本的なスクリーニング法です。
2. 原因遺伝子ABCD1と病態メカニズムの全体像
X-ALDのすべての病態の出発点は、X染色体長腕(Xq28)に位置するABCD1遺伝子の病的バリアントです。この遺伝子がコードするタンパク質「ALDP」の機能が失われることで、細胞内でのVLCFAのペルオキシソーム取り込みが障害されます。
💡 用語解説:ABCD1遺伝子とALDPタンパク
ABCD1(ATP-binding cassette transporter subfamily D member 1)遺伝子は、ペルオキシソーム膜に存在するトランスポーター(運搬役)タンパク質「ALDP(副腎白質ジストロフィータンパク)」をコードしています。ALDPはVLCFAをペルオキシソームの中へ運び込む”関所”の役割を果たします。この関所が壊れると、VLCFAが細胞内に溢れ出し、全身に蓄積して毒性を発揮します。現在までに940種類以上の病的バリアントが報告されています。
💡 用語解説:ペルオキシソームとは
細胞の中にある小器官のひとつで、脂肪酸の分解(β酸化)や有害物質の処理を担います。ミトコンドリアと協力して細胞のエネルギー代謝を支えています。X-ALDではペルオキシソームでのVLCFA分解が機能しなくなることで、全身の細胞に脂肪酸が溜まり続けます。
二元的な病態カスケード:脳の「炎症」と脊髄の「変性」
VLCFAの蓄積から始まるX-ALDの病態は、大きく2つの独立したプロセスに分岐します。ひとつは大脳における激烈な炎症性脱髄(CALD)、もうひとつは脊髄における緩やかな軸索変性(AMN)です。この2つは発症メカニズムが根本的に異なるため、治療戦略も全く別物となります。
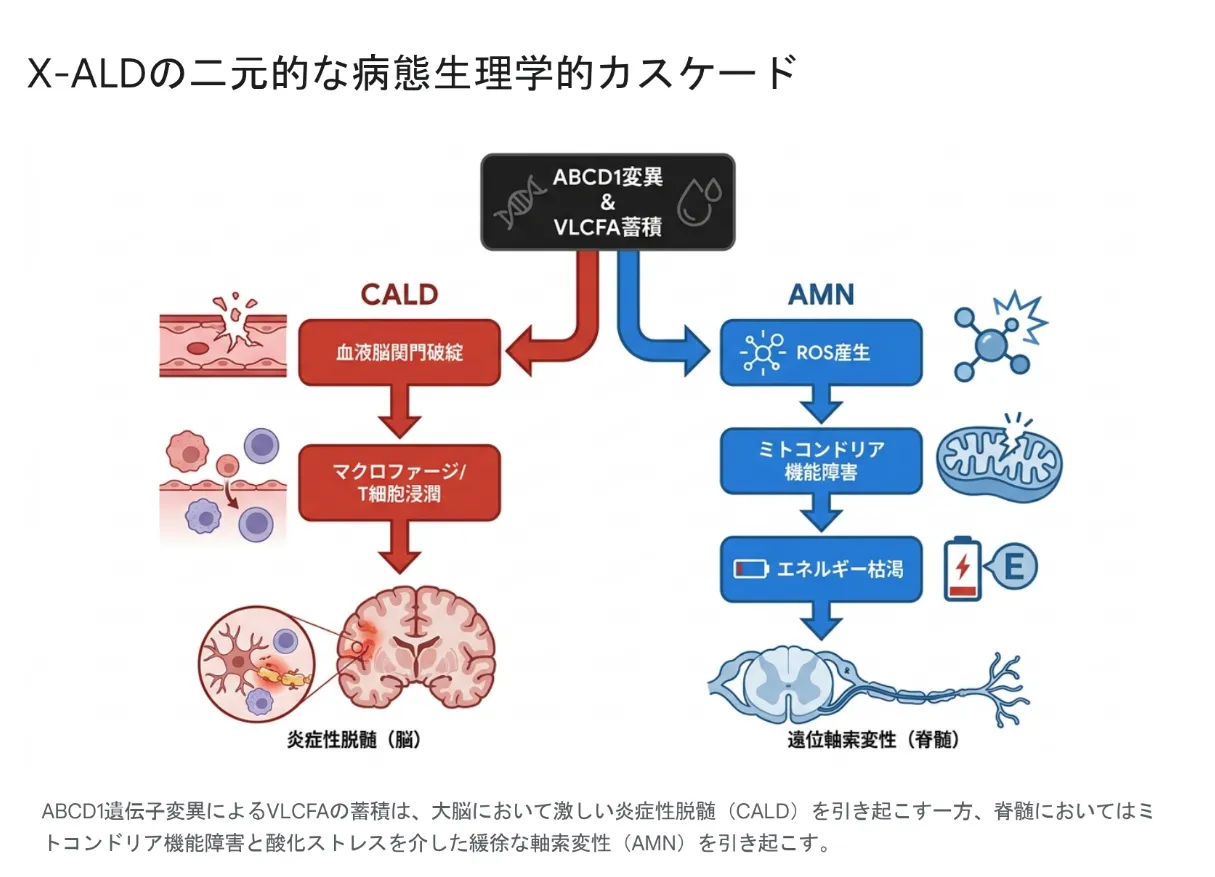
ABCD1遺伝子変異によるVLCFAの蓄積は、大脳では血液脳関門の破綻を介した激しい炎症性脱髄(CALD)を、脊髄ではミトコンドリア機能障害と酸化ストレスを介した緩徐な軸索変性(AMN)を引き起こす。
AMNの病態:酸化ストレスとミトコンドリアエネルギー枯渇
AMNの主な病理は、脊髄の皮質脊髄路と後索における「Dying-back現象」です。神経線維の遠い末端から細胞体に向かって逆行性に変性していく現象で、数十年をかけて緩やかに進行します。重要なのは、AMNの初期には脳の炎症(CALD)は認められないという点です。
💡 用語解説:酸化ストレスとミトコンドリア機能障害
細胞内に蓄積したVLCFAは、活性酸素種(ROS)と呼ばれる有害物質を大量に発生させます。これが「酸化ストレス」です。同時に、VLCFAはエネルギー産生工場であるミトコンドリアの機能も障害し、ATP(細胞のエネルギー通貨)の産生を著しく低下させます。脊髄の長い神経線維はエネルギーを大量に消費するため、このエネルギー枯渇に特に脆弱で、遠位部から徐々に機能を失っていきます。
CALDの病態:血液脳関門の破綻と激しい神経炎症
大脳型(CALD)では、VLCFAによる酸化ストレスが一定の閾値を超えると、突然血液脳関門が破綻します。本来は免疫細胞が入れないはずの脳内にマクロファージやCD8陽性T細胞が大量流入し、激烈な自己免疫性の脱髄反応が連鎖的に引き起こされます。これは放置すれば数年で植物状態に至る、極めて破壊的なプロセスです。
💡 用語解説:血液脳関門(BBB)とは
脳は非常に繊細な臓器であるため、血液中の物質が自由に脳内へ入り込まないよう守るバリアがあります。これが血液脳関門(Blood-Brain Barrier: BBB)です。通常、免疫細胞やウイルス、細菌はこのバリアによって遮断されます。CALDではVLCFAがこのバリアを傷つけ、免疫細胞が脳内に侵入して神経組織を攻撃します。成人AMN患者の約20〜30%がCALDへ移行するリスクを生涯にわたって抱えています。

3. 主な症状と病型スペクトラム
X-ALDの臨床像は、発症年齢・性別・どの臓器が主に侵されるかによって大きく異なります。主な病型は以下の4つに分類されますが、同一患者が複数の病型特徴を持つこともあります。
① 小児大脳型副腎白質ジストロフィー(cCALD)
X-ALDの中で最も重篤かつ進行が早い病型です。典型的には4歳から10歳の男児に発症します。発症初期は多動・学習障害・注意力の低下など、ADHDに似た非特異的な症状を呈するため早期診断が困難です。
🔴 初期症状(4〜10歳)
- 多動・集中力低下(ADHDに類似)
- 学習障害・成績の急激な低下
- 行動の変化・感情の不安定
🔵 進行後の症状
- 進行性の認知機能低下・失語
- 視力障害・難聴
- 重度の痙性麻痺・歩行不能
- 嚥下機能の喪失
無治療の場合、症状発現から2〜3年以内に完全な植物状態に至るか、呼吸器合併症により死亡するという過酷な自然史を辿ります。11歳〜21歳の青年期、さらには成人期に初めてCALDを発症する患者も存在します。
② 成人発症型副腎脊髄ニューロパチー(AMN)
AMNは、X-ALDの中で最も多くの患者が経験する病型です。cCALDを発症しなかったほぼすべての男性患者が、最終的にこの形態に移行します。男性では20〜40歳が発症のピークです。
💡 用語解説:AMN(副腎脊髄ニューロパチー)の3大症状
① 進行性痙性対麻痺:下肢のこわばり(痙縮)・筋力低下・ぎこちない歩行が徐々に進行します。脊髄の長い神経線維の遠位部から変性する「遠位性軸索ニューロパチー」が原因です。
② 自律神経障害:神経因性膀胱・神経因性腸管(便秘・失禁)・勃起障害が早期から現れ、生活の質を著しく損ないます。
③ 感覚障害・神経障害性疼痛:下肢の振動覚・固有受覚の低下と、焼けるような神経障害性疼痛が持続します。
AMNは進行が非常に緩慢であるため、多発性硬化症や他の遺伝性脊髄疾患と誤診されるケースが後を絶ちません。また、AMN患者の約20〜30%は経過中に大脳型(CALD)へ移行するリスクを抱えており、定期的なMRI監視が不可欠です。
③ 女性保因者における発症:2024年のパラダイムシフト
かつて女性は「無症候性の保因者」と考えられていましたが、これは現在では完全に否定されています。ABCD1遺伝子の病的バリアントを持つ女性の少なくとも半数が、加齢とともに確実にミエロパチー(脊髄症)を発症します。女性の場合は発症が遅く、40歳〜65歳頃に歩行障害・感覚障害・神経障害性疼痛が現れることが多いです。
2024年の国際ガイドラインにより、女性保因者は「患者」として積極的な神経学的モニタリングと症状管理の対象とすることが正式に位置づけられました。女性はCALDや重篤な副腎不全を発症することは稀ですが、AMN様症状への介入と長期フォローアップが推奨されています。
④ 原発性副腎皮質機能低下症(Addison病型)
神経症状とは独立して、X-ALD男性患者の多くは早期から副腎皮質機能低下症を発症します。最も一般的には7.5歳までに何らかの兆候が現れます。口腔粘膜の色素沈着・慢性的な疲労感・原因不明の嘔吐・体重減少などが特徴的で、重大なストレス下では生命を脅かす「副腎クリーゼ」を引き起こします。
⚠️ 重要:副腎クリーゼとは
副腎から十分なホルモンが分泌されない状態で感染症・手術・外傷などの強いストレスが加わると、急激な血圧低下・意識障害・嘔吐・循環不全に陥り、生命の危機となります。早期からのグルココルチコイド・ミネラルコルチコイド補充療法が必須です。ただし、ホルモン補充療法は副腎不全をコントロールできても、神経系の進行(AMNやCALD)を予防する効果はありません。
4. 鑑別すべき疾患との違い
AMNは症状が緩徐に進行するため、他の疾患と混同されやすく、診断までに数年〜十数年かかることも珍しくありません。以下の疾患との鑑別が特に重要です。
多発性硬化症(MS)との鑑別
共通点:脊髄症・歩行障害・MRIでの白質病変
鑑別点:AMNでは再発寛解パターンがなく、血漿VLCFA・C26:0-LPC高値、ABCD1遺伝子変異で確定。MSでは通常女性に多く副腎不全を合併しない。
遺伝性痙性対麻痺(HSP)との鑑別
共通点:緩徐進行性の下肢痙縮・歩行障害
鑑別点:AMNでは副腎機能低下・血漿VLCFA上昇・性連鎖遺伝パターン(男性に重症例多)が特徴。HSPは原因遺伝子が多様(SPG遺伝子群)。
原発性副腎不全(他の原因)との鑑別
共通点:副腎皮質機能低下・疲労・色素沈着
鑑別点:特に男児の原因不明の副腎不全ではX-ALDを積極的に除外する必要あり。血漿VLCFA測定が第一選択。自己免疫性副腎炎(抗21-水酸化酵素抗体)との鑑別も重要。
電気生理学的検査(神経伝導検査)では、AMNでは近位部の運動神経伝導速度の著しい低下は認められず、遠位筋の複合筋活動電位(CMAP)振幅が特異的に低下します。これは一次性の脱髄性多発神経炎(急性炎症性脱髄性多発神経炎など)とは明確に区別される所見です。
5. 診断方法・遺伝子検査・長期モニタリング
生化学的診断:血漿VLCFAとC26:0-LPC
X-ALDが疑われる場合、最初のステップは血液検査による生化学的スクリーニングです。男性患者ではほぼ100%の感度・特異度で異常が検出できます。
🧪 男性の第一選択検査
血漿中のVLCFAパネル:
・ヘキサコサン酸(C26:0)濃度
・C24:0/C22:0比
・C26:0/C22:0比
これらの上昇が確認されれば確定診断に向かう第一歩となります。
🧪 女性には特別な検査が必要
女性(ヘテロ接合体)の約20%は血漿VLCFA値が正常範囲内に留まるという重大な落とし穴があります。そのため2024年ガイドラインではC26:0-LPC測定が強く推奨されています。
💡 用語解説:C26:0-LPC(C26:0-リゾホスファチジルコリン)とは
VLCFAが結合したリゾホスファチジルコリンという物質で、X-ALD患者では血中に著明に増加します。従来の血漿VLCFAパネルよりも感度・特異度が高く、女性保因者でも安定して検出できる最新のバイオマーカーです。新生児スクリーニングでも乾燥ろ紙血(DBS)を用いたMS/MS法でC26:0-LPCを測定する方法が標準になっています。
ABCD1遺伝子検査による確定診断
生化学的検査で異常が認められたら、末梢血を用いたABCD1遺伝子の塩基配列解析(DNAシーケンス)で確定診断を行います。940種類以上の病的バリアントが登録されていますが、「意義不明バリアント(VUS)」が検出された場合は、培養皮膚線維芽細胞を用いた機能解析などで病的意義を慎重に判断する必要があります。
Loesスコア:MRI評価の国際標準指標
💡 用語解説:Loesスコアとは
1994年に提唱された脳MRIの国際標準評価指標(34点満点)です。脱髄病変の解剖学的部位・広がり・脳萎縮の程度を数値化します。各領域で正常=0点、片側性病変=0.5点、両側性病変・萎縮=1点と加算していきます。Loesスコア「10未満」かつ神経学的機能スコア(NFS)が0〜1の段階でHSCTや遺伝子治療を行うことが、神経機能の温存に不可欠な条件とされています。
| 評価対象の脳領域 | 最大スコア | 特徴・臨床的意義 |
|---|---|---|
| 頭頂後頭部白質 | 4点 | 患者の約80%がこの領域から発症する初期病変部位 |
| 前頭葉白質 | 4点 | 約15%の患者が前頭葉から発症する「前方優位型」 |
| 視覚路 | 4点 | 視放線・外側膝状体の脱髄を評価、視覚障害リスク判定 |
| 聴覚路 | 4点 | 脳幹を通る聴覚伝導路の脱髄・聴覚障害進行予測 |
| 全般性萎縮 | 4点 | 脳全体の容積減少を軽度〜重度の段階で評価 |
| その他(基底核・脳幹・小脳・内包など) | 14点 | 痙性麻痺の予後判定や深部灰白質障害を総合評価 |
新生児スクリーニング(NBS)の革新
💡 用語解説:新生児スクリーニング(NBS)とは
生まれたばかりの赤ちゃんのかかとから採血した乾燥ろ紙血(DBS)を用いて、発症前に先天性疾患を検出する公的スクリーニング制度です。X-ALDは2013年に米国ニューヨーク州で初めてNBS対象疾患となり、2016年には米国の推奨統一スクリーニングパネル(RUSP)に正式追加されました。タンデム質量分析(MS/MS)法でC26:0-LPCを測定します。NBSにより、副腎クリーゼの予防と、CALDの最適なタイミングでの治療介入が可能になっています。
2024年ガイドライン:患者群別の長期サーベイランス
| 対象患者群 | モニタリング項目 | 推奨頻度・目的 |
|---|---|---|
| 全男性患者 | 副腎機能評価(コルチゾール・ACTH・レニン) | 定期的実施/致死的副腎クリーゼの予防 |
| CALD未発症の男性患者 | 頭部MRI(造影あり・なし) | 年1回以上継続/CALDへの移行を早期捕捉 |
| 全AMN患者(男女) | 神経学的評価(歩行・振動覚・固有受覚) | 定期的実施/脊髄症の進行定量化・対症療法の最適化 |
| 全AMN患者 | 脊髄MRI | 診断時のベースライン撮影のみ推奨。新たな臨床的懸念時に随時実施 |
6. 治療と長期管理の最前線
ロレンツォのオイル:現在は非推奨
映画でも知られる「ロレンツォのオイル」(エルカ酸とオレイン酸を用いた食事療法)は、血漿VLCFA値を下げる効果は確認されました。しかし、その後の厳密な長期臨床研究においてAMNの脊髄変性の進行を止める効果も、CALDへの移行を予防する効果も示されませんでした。2024年の最新ガイドラインでは、日常的な使用は推奨されていません。
造血幹細胞移植(HSCT):早期CALDへの唯一の確立した手段
💡 用語解説:造血幹細胞移植(HSCT)とは
ドナーから提供された正常な造血幹細胞を移植することで、正常な免疫細胞・骨髄細胞を再構築する治療法です。X-ALDでは、移植後にドナー由来の細胞が脳内に定着し、蓄積したVLCFAを分解するとともに神経炎症を鎮める効果があります。ただし大脳の炎症(CALD)にしか有効ではなく、AMNの脊髄変性や副腎機能低下には効果がありません。また「タイミングがすべて」で、Loesスコア10未満・NFS 0〜1・内包両側病変なしの段階での移植が条件です。
遺伝子治療 Skysona(エリバルドジーン オートテムセル):光と影
HSCTの最大の問題は「適合ドナーを探す時間の喪失」と「GVHD(移植片対宿主病)による致死リスク」でした。これを克服すべく開発されたのが、患者自身の造血幹細胞を用いた体外(エクスビボ)遺伝子治療「Skysona(elivaldogene autotemcel)」です。
💡 用語解説:遺伝子治療(エクスビボ遺伝子治療)とは
患者自身の造血幹細胞(CD34+細胞)を体から取り出し(エクスビボ)、レンチウイルスベクターと呼ばれる運搬ツールを使って正常なABCD1遺伝子を細胞に組み込み、その細胞を患者に戻す一回限りの治療です。自己細胞を使うためGVHDのリスクがなく、適合ドナー不要という大きな利点があります。2022年に米国FDAが4〜17歳の早期活動性CALD患者に承認しました。
⚠️ 2024〜2025年最新動向:深刻な発がんリスク
2024年11月にFDAが重大な安全性通信を発表。レンチウイルスベクターのゲノム挿入に関連して、骨髄異形成症候群(MDS)・急性骨髄性白血病(AML)などの造血器腫瘍が発生するリスクが急増していることが判明しました。2025年7月時点で、臨床試験参加者67名中10名(約15%)に血液がんが発生。当初承認時の約4%から3倍以上に増加しています。現在FDAはSkysonaの適応を「適切な適合ドナーが存在しない患者」に限定し、治療後最低15年間の厳重な血液学的モニタリングを義務付けています。
新規疾患修飾薬 レリグリタゾン(Leriglitazone):最大の希望
AMN・CALDの双方に対する最も有力な新規治療薬として開発の最終段階にあるのが、レリグリタゾン(開発コード:NEZGLYAL)です。血液脳関門を通過し、脳・脊髄の両方に作用する低分子化合物です。
💡 用語解説:PPAR-γ(ペルオキシソーム増殖因子活性化受容体ガンマ)とは
細胞の核内にある転写因子(遺伝子のスイッチをオン・オフする調節タンパク)のひとつです。PPAR-γを活性化すると、ミトコンドリアの生合成機能が回復し、酸化ストレスが軽減され、ミクログリアやアストロサイトの過剰な炎症反応が抑制されます。レリグリタゾンはこのPPAR-γを選択的に活性化することで、AMNの軸索変性とCALDの炎症性脱髄の両方にアプローチする画期的なメカニズムを持ちます。
ADVANCE試験(成人AMN患者・第2/3相)の結果
歩行障害を有する成人男性AMN患者116名を対象にした2年間の国際二重盲検プラセボ対照試験です。主要評価項目(6分間歩行テストの変化量)では統計的に有意な差に到達しませんでした。しかし、最も重要な発見は副次的なデータから生まれました——試験期間中にCALDへ移行したのはプラセボ群6名、レリグリタゾン群は0名(p=0.0015)。さらに、脳のLoesスコア上昇と神経損傷バイオマーカー(血中NfL)も有意に抑制されました。AMNの歩行症状を即座に治すわけではないが、「最も恐ろしいCALDへの移行」を強力に予防する神経保護薬として機能することが示されました。
NEXUS試験(小児cCALD・第2/3相):病態進行停止の画期的データ
活動性の初期cCALDを有する小児男児を対象にした試験です。2024〜2025年に報告されたデータは極めて有望なものでした。
📊 NEXUS試験:cCALD病態進行停止率の比較(24週時)
主要評価項目:Arrested Disease(病態進行停止)達成率
(推定停止率)
治療群
p < 0.05(自然停止率を統計学的に有意に上回る)
データソース:NEXUS試験 (PMC12159931) / Minoryx Therapeutics
これらの強力なエビデンスを基盤として、開発企業(Minoryx Therapeutics社・Neuraxpharm社)は2025年半ばを目標にレリグリタゾンの製造販売承認申請(MAA)を欧州医薬品庁(EMA)に提出し、現在審査が進行中です。承認されればX-ALDにおける初の経口疾患修飾薬となり、治療の選択肢が大きく広がります。
AMNの包括的な対症療法
現時点(2026年)でAMNの軸索変性を完全に止める承認薬はなく、多職種チームによる対症療法が治療の主軸です。
🚽 神経因性腸管・膀胱の管理
- 段階的アプローチ(食事・水分・薬物・経肛門的洗腸療法)
- 清潔間欠導尿(CIC)の導入
- 膀胱壁内ボツリヌス毒素注射
💊 痙縮・疼痛管理
- バクロフェン(GABA-B受容体作動薬)内服
- ボツリヌス毒素局所注射
- 重症例:バクロフェン髄腔内投与(ITB療法)
- 神経障害性疼痛:プレガバリン・デュロキセチン・三環系抗うつ薬
🏃 リハビリ・副腎管理
- 理学療法・作業療法による機能維持
- 歩行補助具・車いすの適切な導入
- グルココルチコイド・ミネラルコルチコイド補充療法(副腎不全)
D2受容体拮抗薬(一部の制吐薬・抗精神病薬)の使用は、白質ジストロフィー患者では遅発性ジスキネジア等の運動障害を誘発するリスクがあるため厳格に避けるべきです(2021年ロイコジストロフィー管理コンセンサスガイドライン)。
7. 遺伝カウンセリングと家族計画
X-ALDの確定診断後、患者本人と家族に対して丁寧な遺伝カウンセリングが行われます。臨床遺伝専門医が遺伝の仕組み、再発リスク、検査の選択肢を一緒に考えます。
- ➤遺伝形式の理解:母親が保因者の場合、息子は50%の確率で発症し、娘は50%の確率で保因者となります。父親が患者の場合、息子には遺伝しませんが、娘は全員が保因者となります。
- ➤保因者検査の重要性:女性保因者は自分では気づいていないことが多く、拡張型キャリアスクリーニングや保因者検査を通じて自身の状態を把握することが、本人の健康管理と次世代への対応において非常に重要です。
- ➤妊娠中の選択肢:既知の変異が家族内にある場合、羊水検査・絨毛検査による出生前診断が可能です。また、着床前遺伝学的検査(PGT)の適応についても専門医にご相談ください。
- ➤家族全体へのアプローチ:患者の兄弟・おじ・男性の甥などもリスクのある血縁者として、スクリーニングの対象となります。診断が明確になることで、副腎クリーゼの予防や早期MRIモニタリングなど、具体的な命を守る行動につながります。
8. よくある誤解
誤解①「男の子だけの病気」
X連鎖劣性遺伝のため男性に重症例が多いのは事実ですが、女性保因者の50%以上が人生のうちにAMNを発症します。女性も当事者として積極的なモニタリングが必要です(2024年ガイドライン)。
誤解②「保因者は発症しない」
「保因者=無症候性」は古い概念です。女性保因者のVLCFA値が正常でも安心してはいけません。C26:0-LPC検査により正確な診断が可能で、定期的な神経学的フォローが重要です。
誤解③「変異の種類でどの病型になるかわかる」
現在の医学では遺伝子型から将来の病型を予測することは不可能です。同じ変異を持つ兄弟が全く異なる病型を呈することもあります。これがX-ALDの最大の謎であり、定期的な監視が不可欠な理由です。
誤解④「ロレンツォのオイルで治る」
血液のVLCFA値は下がりますが、AMNの進行を止める効果もCALDへの移行を予防する効果も臨床的に証明されていません。2024年の最新ガイドラインでは日常的な使用は推奨されていません。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 X-ALD・AMN・保因者について相談したい方へ
副腎白質ジストロフィーの診断・保因者検査・遺伝カウンセリングのご相談は
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にどうぞ。
参考文献
- [1] GeneReviews. X-Linked Adrenoleukodystrophy. NCBI Bookshelf. [NBK1315]
- [2] Adrenoleukodystrophy. StatPearls. NCBI Bookshelf. [NBK562328]
- [3] Mitochondrial Dysfunction and Impaired Oxidative Stress Defense as Potential Trigger of Cerebral X-linked Adrenoleukodystrophy. PubMed 2025. [PMID: 41177236]
- [4] Newborn Screening for X-Linked Adrenoleukodystrophy: Past, Present, and Future. PMC. [PMC8884000]
- [5] Pathophysiology of X-Linked Adrenoleukodystrophy: Updates on Molecular Mechanisms. PMC. [PMC11271253]
- [6] Practical Approach to Longitudinal Neurologic Care of Adults With X-Linked Adrenoleukodystrophy. Neurology: Genetics 2024. [Neurology Genetics]
- [7] Prognostication of X-Linked Adrenoleukodystrophy Based on the Loes Neuroimaging Score. Neurology 2023. [Neurology]
- [8] SKYSONA Gene Therapy for Cerebral Adrenoleukodystrophy. Boston Children’s Hospital. [Boston Children’s Hospital]
- [9] FDA Approves Required Labeling Changes for Increased Risk of Hematologic Malignancy Following Treatment with Skysona. FDA 2024. [FDA.gov]
- [10] Safety and efficacy of leriglitazone for preventing disease progression in men with adrenomyeloneuropathy (ADVANCE): a randomised, double-blind, multi-centre, placebo-controlled phase 2-3 trial. PubMed 2023. [PMID: 36681445]
- [11] Safety and efficacy of leriglitazone in childhood cerebral adrenoleukodystrophy (NEXUS): an interim analysis of an open-label, phase 2/3 trial. PMC 2025. [PMC12159931]
- [12] Newborn Screening for X-linked Adrenoleukodystrophy – RUSP Addition. HRSA 2018. [HRSA.gov]






