目次

📍 クイックナビゲーション
LMBRD1遺伝子は、第6染色体長腕(6q13)に位置し、リソソーム膜タンパク質LMBD1をコードする遺伝子です。ビタミンB12(コバラミン)をリソソームから細胞質へと送り出す際の「エスコート役」として機能することで広く知られていますが、近年の研究ではそれにとどまらず、インスリン受容体の細胞内取り込み制御・D型肝炎ウイルスの核外輸送・初期胚発生の原腸陥入開始にも不可欠であることが明らかになっています。両アレルに機能喪失変異が生じると、致死的にもなりうる先天性代謝異常症「メチルマロン酸血症・ホモシスチン尿症cblF型」を引き起こします。さらに2024〜2025年には、角化不全症に類似した新規表現型の報告や、パーキンソン病の新規リスク遺伝子座としての確立が相次いで報告され、基礎・臨床遺伝学の両分野から熱い視線が注がれています。
Q. LMBRD1遺伝子とはどのような遺伝子ですか?まず結論だけ知りたいです
A. 第6染色体(6q13)に位置し、リソソーム膜に局在するLMBD1タンパク質をコードする遺伝子です。ビタミンB12(コバラミン)のリソソームから細胞質への輸送複合体における必須エスコートタンパク質として機能し、機能喪失型変異はcblF型メチルマロン酸血症・ホモシスチン尿症という重篤な先天性代謝異常症を引き起こします。単なるビタミントランスポーターの補助タンパク質にとどまらない多面的機能が近年明らかになっています。
- ➤遺伝子の基本情報 → 6q13、17エクソン、LMBD1(533アミノ酸、60.6 kDa)、25以上の組織でユビキタス発現
- ➤多面的な分子機能 → ①ビタミンB12ベクトル型輸送、②インスリン受容体エンドサイトーシス制御、③NESIアイソフォームによるHDV核外輸送、④原腸陥入制御
- ➤cblF型の病態 → リソソーム内B12蓄積 → MMA・ホモシステイン蓄積 → 多臓器不全(血液・神経・消化器系)
- ➤最新知見(2024-2025) → 角化不全症様の新規表現型・パーキンソン病GWAS新規リスク遺伝子座に同定
- ➤診断と治療 → MMA・tHcy同時上昇が決定的バイオマーカー、ヒドロキソコバラミン大量補充療法が治療の根幹
1. LMBRD1遺伝子とは:ゲノム構造・発現・タンパク質
LMBRD1(LMBR1 domain containing 1)遺伝子は、ヒト第6染色体長腕(6q13)の69,674,010〜69,797,010塩基対の領域に位置する、計17エクソンからなるタンパク質コード遺伝子です。遺伝子ID(NCBI)は55788であり、LMBD1・NESI・MAHCF・C6orf209など複数の別名を持ちます。コードされる主要タンパク質はLMBD1(lysosomal cobalamin transport escort protein LMBD1)と呼ばれ、533アミノ酸、分子量約60.6 kDaの膜タンパク質です。
💡 用語解説:LIMRファミリー
LMBD1は進化的によく保存されたLIMRファミリー(LMBRD1サブファミリー)に属します。LIMRファミリーは多回膜貫通型タンパク質の一群で、脂質やビタミンなどの疎水性・両親媒性小分子の膜輸送に関わる構造的特徴(複数の膜貫通ドメイン)を持ちます。LMBD1は9つの膜貫通ドメイン(TMD)とコイルドコイルドメイン(アミノ酸残基234〜289)を有する多回膜貫通型タンパク質であることが生化学的解析によって確認されています。
発現パターン:25以上の組織でユビキタスに発現
RNAシーケンシング(RNA-Seq)などのトランスクリプトーム解析によれば、LMBRD1遺伝子は副腎(RPKM 45.7)・腎臓(RPKM 42.7)をはじめ、少なくとも25以上の全身組織においてユビキタスに発現しています。この広範な発現パターンは、LMBD1タンパク質が特定の臓器に特化した機能を担うのではなく、あらゆる真核細胞の生存と恒常性維持に不可欠な基礎的プロセス(ハウスキーピング機能)を担っていることを強く示唆しています。
💡 用語解説:ハウスキーピング遺伝子
ほぼすべての細胞種・組織においてほぼ一定に発現し続ける遺伝子の総称です。細胞の生存・増殖・代謝などの基本的な機能を維持するために常に必要とされます。LMBRD1がハウスキーピング遺伝子的な発現パターンを示すことは、その機能が特定の代謝経路の補助にとどまらず、細胞膜系全体のトポロジー制御という根本的な生物学的プロセスに関与していることを意味します。
LMBD1の三次元構造と相互作用ネットワーク
最新の構造解析(Blue Native PAGE・化学架橋法・SEC-MALSなど)により、界面活性剤で可溶化されたLMBD1は単量体ではなくホモ二量体(ホモダイマー)を形成することが実証されています。さらに表面プラズモン共鳴(SPR)法を用いたリアルタイム結合親和性解析によって、LMBD1は以下の重要なタンパク質と極めて強い結合力(低ナノモルレベル)を持つことが明らかになりました。
🔗 ABCD4タンパク質
同じくリソソーム膜に局在するABCトランスポーター。低ナノモルの強い親和性でLMBD1と結合し、ビタミンB12を通過させるポア(孔)そのものを形成する。LMBD1はABCD4を小胞体からリソソームへと正しく局在させるシャペロン的役割も担う。
🔗 MMACHCタンパク質
細胞質側でビタミンB12を受け取り、その後の代謝経路へと誘導するコバラミン処理タンパク質。LMBD1は MMACHCをリソソーム膜の極近傍にリクルートし、リソソームから排出されたB12を直接手渡す「ベクトル型輸送」を実現する。
これらの結合データは、LMBD1が単独で浮遊しているのではなく、LMBD1・ABCD4・MMACHCの三者が物理的に強固な複合体(マシナリー)を形成し、高度に制御された分子チャネルとして機能することを生化学的に裏付けています。
2. LMBD1タンパク質の多面的な分子機能
LMBRD1遺伝子の最大の細胞生物学的特徴は、その多面発現性(Pleiotropy)にあります。翻訳産物であるLMBD1タンパク質は、細胞内の全く異なる局在(リソソーム膜・細胞膜・核周囲)において、完全に独立した生物学的プロセスのアダプターとして機能します。現在明らかになっている主な機能は以下の4つです。
💡 用語解説:多面発現性(Pleiotropy)
1つの遺伝子・タンパク質が、一見全く無関係に見える複数の生物学的プロセスや表現型(形質)に影響を与える性質のことです。LMBD1はリソソーム・細胞膜・核周囲という3つの異なる細胞内コンパートメントで異なる相手と結合し、異なる機能を果たすという、真核細胞の複雑な生命制御の典型例を示しています。
①リソソームにおけるビタミンB12のベクトル型輸送
LMBD1の最も古典的かつ詳細に解明されている機能は、細胞内コバラミン(ビタミンB12)代謝における必須のトランスポーター複合体形成です。食事から摂取されたビタミンB12はトランスコバラミンと結合し、受容体媒介性エンドサイトーシスによって細胞内に取り込まれ、最終的にリソソームへと運ばれます。
💡 用語解説:コバラミン(ビタミンB12)
コバルト原子を中心に持つ水溶性ビタミンの総称です。ヒトの細胞内では主に2つの活性型補酵素(アデノシルコバラミン:AdoCbl と メチルコバラミン:MeCbl)として機能します。AdoCblはミトコンドリアでメチルマロニルCoAムターゼの補酵素として、MeCblは細胞質でメチオニン合成酵素の補酵素として、それぞれ不可欠な役割を担います。分子量が大きく水溶性であるため、リソソーム膜を単純拡散で通過できず、専用の輸送機構が必要です。
このビタミンB12輸送において、LMBD1は2段階の役割を担います。第1段階として、小胞体で合成されたABCD4トランスポーターをリソソーム膜へと正確にトランスロケーションさせるシャペロン的機能を発揮します。LMBRD1をノックアウトした細胞実験では、ABCD4がリソソームに到達できず小胞体内に滞留することが確認されています。
第2段階として、リソソーム膜上でLMBD1はABCD4との機能的ヘテロ複合体を形成するとともに、細胞質に存在するMMACHCをリソソーム膜の極近傍にリクルートします。ビタミンB12を物理的に通過させるポアはABCD4がATPエネルギーを利用して形成しますが、生体内ではABCD4単独ではこのプロセスは完結しません。LMBDのMMACHCリクルートにより、リソソームから排出された直後のビタミンB12が細胞質内で希釈されることなく、直接MMACHCへと手渡される——これが「ベクトル型(方向性を持った)輸送」と呼ばれる精巧な機構です。
🔬 LMBD1-ABCD4-MMACHC複合体によるビタミンB12ベクトル型輸送のメカニズム
食事から
ビタミンB12
エンドサイトーシス
リソソーム内に取込
LMBD1+ABCD4複合体
リソソーム膜を通過
MMACHCへ直接受け渡し
細胞質へ
ミトコンドリア経路
AdoCbl → メチルマロニルCoAムターゼの補酵素
細胞質経路
MeCbl → メチオニン合成酵素の補酵素
cblF型ではLMBD1機能不全によりリソソーム内にB12が滞留し、両経路が停止する
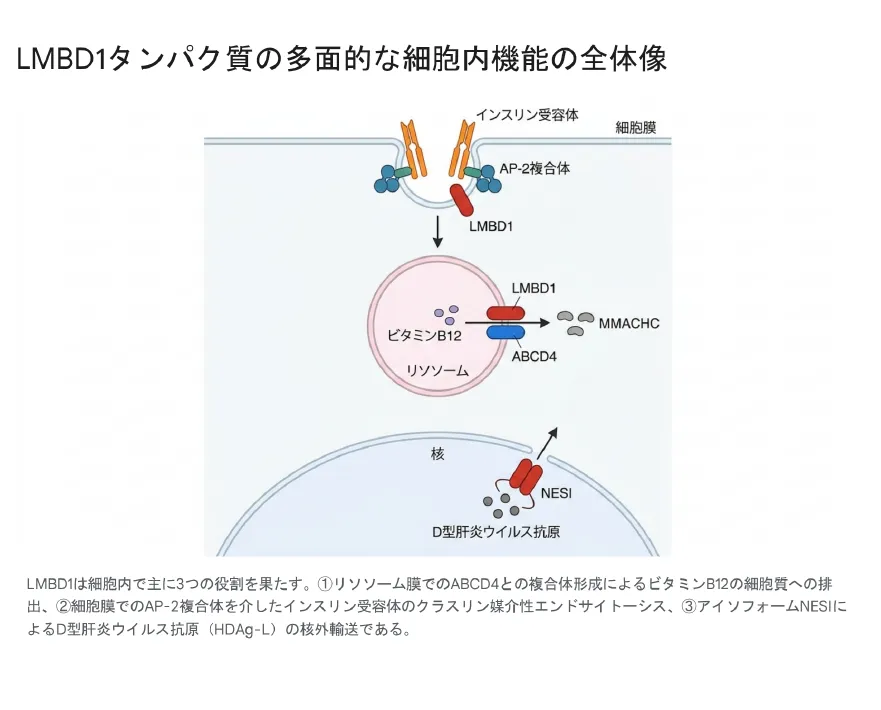
LMBD1は細胞内で主に3つの役割を果たす。①リソソーム膜でのABCD4との複合体形成によるビタミンB12の細胞質への排出、②細胞膜でのAP-2複合体を介したインスリン受容体のクラスリン媒介性エンドサイトーシス、③アイソフォームNESIによるD型肝炎ウイルス抗原(HDAg-L)の核外輸送。
②インスリン受容体(INSR)のエンドサイトーシスと糖代謝制御
LMBD1はリソソームに局在するだけでなく、細胞の原形質膜においてインスリン受容体(INSR)の細胞内取り込みを仲介する特異的アダプターとしても機能します。インスリンがINSRに結合してチロシンキナーゼシグナルが活性化された後、そのシグナルをオフにするためにINSRはクラスリン媒介性エンドサイトーシスによって速やかに細胞内へと回収される必要があります。
💡 用語解説:クラスリン媒介性エンドサイトーシス
細胞膜上の受容体やリガンドを細胞内に取り込む主要な経路のひとつです。クラスリンと呼ばれるタンパク質が膜の内側に格子状の被覆(coat)を形成し、膜を内側に陥入させて小胞を形成することで、受容体を細胞内に取り込みます。アダプタータンパク質複合体2(AP-2)はこの過程で積み荷(受容体)とクラスリンを繋ぐ橋渡し役を担います。LMBD1はYERLおよびWTKFと呼ばれる特定のモチーフを介してAP-2とクラスリン重鎖の両方に直接結合し、INSRをクラスリン被覆小胞へと誘導します。
Lmbrd1遺伝子の発現を人為的に低下させた研究モデルでは、INSRのエンドサイトーシスが顕著に阻害され、受容体が細胞膜上に滞留し続けることで下流のAktシグナルが持続的に活性化され、細胞のグルコース取り込みが異常に上昇することが観察されています。これはLMBRD1がビタミントランスポーターの補助タンパク質としてだけでなく、全身のインスリン感受性や糖代謝ホメオスタシスを制御するシグナルモジュレーターとしての側面も持つことを意味します。
③アイソフォームNESIを介したD型肝炎ウイルス(HDV)の核外輸送
LMBRD1遺伝子は選択的スプライシングまたは異なる翻訳開始メカニズムを通じて、NESI(Nuclear Export Signal-Interacting protein:アイソフォーム3)と呼ばれる別形態のタンパク質も産生します。NESIは細胞膜・リソソームではなく、核と細胞質間の輸送に深く関与します。
NESIはB型肝炎ウイルス(HBV)と共感染するD型肝炎ウイルス(HDV)のライフサイクルにおいて、病原体にハイジャックされる標的となります。HDVの大型デルタ抗原(HDAg-L)内に存在する核外移行シグナル(NES)領域と特異的に相互作用したNESIは、ラミンA/C(核膜の主要構造タンパク質)およびヌクレオポリン(核膜孔複合体タンパク質)と複合体を形成し、一般的な核外輸送受容体であるCRM1とは独立した経路を通じてウイルス・リボ核タンパク質(RNP)の核から細胞質への輸送を促進します。
④初期胚発生と原腸陥入における絶対的な必要性
Cre/LoxPシステムを用いて構築されたLmbrd1完全欠損(ノックアウト)マウスは、着床後の初期胚発生段階において致死となります。ホールマウントin situハイブリダイゼーション(WISH)解析によれば、Lmbrd1−/−胚においてBMP4やNodalなどの近位-遠位軸形成シグナルは正常に機能しているにもかかわらず、その直後に続く「原腸陥入(Gastrulation)」の開始プロセスが著しく破綻していることが確認されています。
💡 用語解説:原腸陥入(Gastrulation)
受精卵が細胞分裂を繰り返した後、細胞の大規模な移動と再配置によって外胚葉・中胚葉・内胚葉という3つの胚葉を形成する発生学的イベントです。この過程が正常に完了することで、脳・脊髄・心臓・腸管・骨格など全身のすべての臓器の原基が確立されます。LMBD1が原腸陥入の開始に必須であるという事実は、インスリン受容体に限らず、発生を誘導する様々な増殖因子受容体の細胞膜上のトラフィッキング全般において、LMBD1が代償不可能な中心的役割を担っていることを示唆しています。

3. cblF型メチルマロン酸血症・ホモシスチン尿症の病態生理と臨床症状
LMBRD1遺伝子の両アレルにおける機能喪失型(Loss-of-Function)変異は、細胞内コバラミン代謝障害の特定サブタイプである「cblF型(cblF defect)」の直接的な原因です。OMIM登録番号は277380、Orphanetには「メチルマロン酸尿症・ホモシスチン尿症cblF型(ORPHA:79284)」として登録されています。数百万人に一人という極めて稀な常染色体潜性(劣性)遺伝疾患ですが、発症した患者には多臓器にわたる深刻な機能不全をもたらします。
💡 用語解説:常染色体潜性(劣性)遺伝
「常染色体」は性染色体(X・Y)以外の染色体、「潜性(劣性)」は2本の染色体の両方に変異がある場合に初めて発症する遺伝形式です。cblF型では、父親由来と母親由来の両アレルに機能喪失変異が存在するときのみ発症します。両親はそれぞれ片方のアレルに変異を持つ「保因者(キャリア)」であり、通常は無症状です。両親が保因者の場合、子どもが発症する確率は理論上25%です。
生化学的メカニズム:二重の代謝ネットワーク崩壊
cblF型の根本的な病態は、LMBD1機能不全によるリソソーム内での遊離コバラミンの異常蓄積(トラップ)です。これにより、細胞質およびミトコンドリアでビタミンB12を補酵素として必要とする2つの重要代謝経路が同時に停止します。
⚡ 第1の破綻:AdoCbl枯渇
ミトコンドリア経路でのアデノシルコバラミン(AdoCbl)が欠乏し、メチルマロニルCoAムターゼが機能停止します。
イソロイシン・バリン・スレオニン・メチオニン・奇数鎖脂肪酸の代謝が停滞し、有毒なメチルマロン酸(MMA)が血中・尿中に大量蓄積。重篤なアシデミアを引き起こす。
⚡ 第2の破綻:MeCbl枯渇
細胞質経路でのメチルコバラミン(MeCbl)が欠乏し、メチオニン合成酵素が機能停止します。
ホモシステインのメチオニンへの再メチル化が停止し、毒性の高い総ホモシステイン(tHcy)が血中に蓄積(ホモシスチン尿症)。同時にメチオニン・SAMの産生が低下する。
💡 用語解説:メチルマロン酸(MMA)とホモシステイン(tHcy)
メチルマロン酸(MMA:Methylmalonic acid)はプロピオン酸代謝の中間産物であり、正常な代謝ではスクシニルCoAへと変換されますが、メチルマロニルCoAムターゼが機能しないと蓄積します。神経毒性・腎毒性を持つ有機酸です。総ホモシステイン(tHcy)はアミノ酸のメチオニンの代謝中間体で、過剰になると血管内皮障害・血栓形成リスクの上昇・神経毒性をもたらします。cblF型の血液検査でこの2つが同時に上昇していることが生化学的診断の決め手となります。
cblF型の主な臨床症状
🩸 血液学的異常
- 巨赤芽球性貧血
- 好中球減少症
- 血小板減少症(汎血球減少)
B12枯渇による「葉酸トラップ」によるDNA合成障害。骨髄など活発に細胞分裂する造血系に最も早期に現れる。
🧠 神経・発達的異常
- 筋緊張低下(フロッピーインファント)
- 重度の精神運動発達遅滞
- 知能障害
- けいれん発作
- 小頭症・進行性脳症
MMAおよびホモシステインの神経毒性、メチル化能低下による髄鞘(マイエリン)形成障害。
🍼 消化器系・全身状態
- 著しい哺乳不良・摂食障害
- 慢性的な成長障害(体重増加不良)
- 頻回な嘔吐
- 無気力状態(レサージー)
全身的なエネルギー代謝不全(TCA回路への基質供給不足)および酸塩基平衡の異常による全身状態の悪化。
🧬 発生学的異常・その他
- 難治性の口内炎・舌炎・皮疹
- 先天性心疾患
- 非定型的な顔貌異常
- 頭蓋骨癒合異常(前頭縫合)
- 片側性腎欠損
LMBD1が胚発生に必須であるという基礎的知見と合致する、発生段階でのモルフォゲン勾配・細胞増殖の乱れ。
4. 変異スペクトルと遺伝子型・表現型相関
LMBRD1遺伝子に確認されている疾患原因変異の大部分は、タンパク質の構造を根本的に破壊するフレームシフト変異やスプライシング異常などの機能喪失型(LoF)変異です。ミスセンス変異の報告は極めて稀であり、これは多回膜貫通型タンパク質であるLMBD1が、わずかなアミノ酸置換であれば全体構造を維持できるか、あるいは逆に胚性致死となって出生に至らないという進化的選択圧の存在を示唆しています。
💡 用語解説:フレームシフト変異
DNAの塩基が1〜2個挿入または欠失することで、コドン(3塩基1組)の読み取り枠(フレーム)がずれる変異です。フレームがずれた後のすべてのアミノ酸配列が本来とは全く異なるものになり、多くの場合、途中に終止コドンが生じて短縮されたタンパク質(機能を持たない)が産生されます。cblF型で最も多い変異c.1056delGはこのフレームシフト変異の典型例です。
最頻変異 c.1056delG(p.L352fsX18)と創始者効果
特に欧米コホートで圧倒的な頻度を占めるのが、エクソン領域における1塩基欠失変異 c.1056delG(p.Leu352fs / p.L352fsX18)です。この変異はフレームシフトを引き起こし早期終止コドンをもたらすため、機能を持たない短縮LMBD1タンパク質が産生されます。報告されているcblF型患者の罹患アレルの約68〜75%がこの単一変異を保有しており、詳細なハプロタイプ解析により共通の1.34メガベース(Mb)ハプロタイプブロック内に存在することが確認され、歴史的な創始者効果によって特定集団内に広まったことが強力に示唆されています。
💡 用語解説:創始者効果(Founder effect)
歴史的に孤立した少数の集団(創始者集団)がより大きな集団に発展したとき、その集団内に特定の遺伝的変異が高頻度に存在する現象です。cblF型のc.1056delGが欧米患者の約70%に見られるのは、この変異を持つ共通の祖先から子孫が広まったことを意味します。遺伝疫学的な研究において「特定地域・民族での有病率を推定する」ための重要な根拠となります。
主要な病原性バリアント一覧
| 変異(cDNA) | タンパク質変化 | 変異タイプ・帰結 | 臨床的背景 |
|---|---|---|---|
| c.1056delG | p.Leu352fs (p.L352fsX18) | 1塩基欠失・フレームシフト・早期終止 | 最頻変異(約70%)。明確な創始者効果あり。ホモ接合体または複合ヘテロ接合体として多数報告。 |
| c.916-1G>T | スプライシング異常 | スプライスアクセプター部位の点変異 | トルコ人患者でc.1056delGとの複合ヘテロ接合体として同定。 |
| c.1339-1G>T | スプライシング異常 | スプライスアクセプター部位の点変異 | トルコ人患者でc.1056delGとの複合ヘテロ接合体として同定。 |
| c.1405delG | p.Asp469fs (p.D469fsX38) | 1塩基欠失・フレームシフト・C末端欠損 | 近親婚のトルコ人患者でホモ接合体として発見。重度の新生児期発症(脳室内出血によるけいれん等)。 |
| c.70-4298_246+2311del6785 | 大規模欠失(エクソン2含む) | 6785塩基対の巨大欠失・完全機能喪失 | c.1056delGとの複合ヘテロ接合体として同定。微小欠失症候群的なメカニズム。 |
| c.562+4_562+7del | エクソン6スキッピング(インフレーム) | イントロン内スプライス部位欠失 | 2025年報告・新規変異。角化不全症様の非典型表現型を引き起こす。ABCD4との親和性低下をモデリングで示唆。 |
コバラミン代謝障害の各cbl型との比較
コバラミン(Cbl)代謝障害は原因遺伝子と障害部位によってcblA型〜cblX型などに分類されます。cblF型の特徴は、MMAとホモシステインの両方が上昇する「複合型」である点であり、MMAのみが上昇する型(cblA・cblB型など)とは異なります。
cblD型
原因遺伝子:MMADHC遺伝子。MMAのみ上昇のサブタイプとMMA・ホモシステイン両方上昇のサブタイプあり。障害部位に依存。
cblJ型
原因遺伝子:ABCD4遺伝子(ABCD4遺伝子ページ)。LMBD1の直接のパートナーであるABCD4の機能喪失。cblF型と病態が極めて近い。
cblC型(二遺伝子性)
MMACHC遺伝子と他遺伝子の複合変異による二遺伝子性(digenic)遺伝形式。単一遺伝子パネルでは見逃しリスクあり。
5. 2024〜2025年の最新研究:新規表現型とパーキンソン病リスク
LMBRD1遺伝子に関する研究は、古典的な先天性代謝異常症の枠を大きく超え、次世代シーケンシング(NGS)と大規模ゲノム疫学データによって全く新しい疾患領域へと展開しています。2024〜2025年に発表された研究は、LMBRD1が関連する臨床スペクトルを劇的に拡大させました。
①角化不全症(Dyskeratosis Congenita)様症状を呈する新規バリアント(2025年)
2025年2月に報告された症例は、LMBRD1遺伝子の特定の新規バリアントが従来のcblF型の枠組みに収まらない複雑な表現型を引き起こすことを初めて明らかにしました。症例は15歳の男性患者であり、全エクソーム解析(WES)の結果、イントロン領域の新規スプライス部位欠失 c.562+4_562+7del をホモまたは複合ヘテロ接合で保有していることが判明しました。cDNA解析でこの変異がエクソン6のスキッピングを引き起こし、インフレームの欠失をもたらすことが確認され、計算機ホモロジーモデリングではABCD4との相互作用親和性の著明な低下が示唆されました。
💡 用語解説:角化不全症(Dyskeratosis Congenita:DC)
DKC1・TERT・TERCなどテロメア維持に関わる遺伝子の変異によって引き起こされる遺伝性疾患です。①爪のジストロフィー(変形・発育不全)、②皮膚の網状の色素沈着、③口腔白板症・粘膜異常の「三主徴」を特徴とし、骨髄不全・肺線維症・悪性腫瘍リスクの上昇を伴います。細胞分裂が活発な造血幹細胞・皮膚・粘膜の基底細胞がテロメア短縮により早期に枯渇することが病態の中心です。
この患者は古典的なcblF症状(発達遅滞・貧血)に加えて、重度の爪のジストロフィー・皮膚の網状色素沈着(reticulated dyschromia)・難治性口腔白板症と口内炎・睾丸萎縮・免疫不全傾向を示しており、これらはまさにDCの古典的三主徴および合併症に他なりません。
なぜビタミンB12輸送障害がテロメア異常症と酷似した症状を引き起こすのか——答えは細胞分裂の依存性にあります。骨髄造血幹細胞・皮膚・粘膜の基底細胞という「活発に細胞分裂を行う上皮系組織」は、DNA合成のために大量のヌクレオチド前駆体を必要とします。LMBD1変異による重篤な細胞内B12枯渇は、テロメアを直接短縮させるわけではありませんが、これらの組織におけるDNA合成・修復能力を致命的に低下させます。その結果として生じる早期の組織枯渇・変性が、臨床的にはDCと見分けのつかない「フェノコピー(phenocopy)」を生み出します。このc.562+4_562+7del変異患者に対してヒドロキソコバラミン(OHCbl)の静脈内投与を行ったところ、著しい皮膚・粘膜症状の改善が見られており、病態の可逆性が示されています。
②パーキンソン病(PD)のGWASにおける新規リスク遺伝子座としての確立(2024-2025年)
Global Parkinson’s Genetics Program(GP2)などが主導した史上最大規模のマルチアンセストリー・メタGWASにおいて、LMBRD1が新規リスク遺伝子として確立されました。この研究は約63,555人のPD患者・17,700人のプロキシケース・1,746,386人の対照群(合計約180万人以上)の遺伝データを統合した未曾有の規模のものです。
💡 用語解説:GWAS(ゲノムワイド関連解析)
GWAS(Genome-Wide Association Study)は、多数の症例と対照群のゲノム全体にわたる多型(SNP)を統計的に比較し、特定の疾患・形質と関連する遺伝的変異を同定する手法です。「メタGWAS」は複数の独立したGWASコホートを統合して解析することで、より高い統計的検出力を持ちます。今回のGP2メタGWASでは134の独立した遺伝子座がPDリスクと有意に関連し、そのうち59が全く新しい新規遺伝子座として同定され、LMBRD1がその中の一つとして確立されました。
重要なのはメカニズムの解釈です。パーキンソン病患者は健常者と比較して血清B12が低い傾向があることは知られていましたが、メンデルランダム化(MR)解析では血清B12レベルの低下そのものがPD発症の直接的な原因とは否定されています。したがってLMBRD1SNPがPDリスクを高めるメカニズムは、全身性B12欠乏によるものではなく、LMBD1本来の「細胞内小胞トラフィッキング」と「リソソーム機能維持」としてのアダプター機能の微細な低下に起因する可能性が高いと考えられます。パーキンソン病の病態中心にはエンドソーム-リソソーム経路の機能不全(GBA遺伝子・LRRK2遺伝子変異が代表例)とα-シヌクレイン凝集タンパク質の蓄積があり、LMBD1の遺伝的変異が長年にわたってリソソームの処理効率をわずかに低下させることで、加齢に伴う神経細胞内での異常タンパク質蓄積リスクを徐々に増大させているという仮説が成立します。
6. 診断プロセスと遺伝子検査
cblF型の診断は生化学的検査による代謝異常の検出と遺伝子検査による変異の同定の2つの柱で構成されます。
生化学的診断:MMAとtHcyの同時上昇が決め手
cblF型の最も重要な生化学的マーカーは、血漿総ホモシステイン(tHcy)の著明な上昇と、血中および尿中メチルマロン酸(MMA)の上昇が同時に観察されることです。この「両方の同時上昇」がcblF型(およびcblC・J型など他の複合型)を示す決定的な生化学的シグネチャーです。
⚠️ 重要な注意点:血清ビタミンB12濃度が正常あるいは軽度上昇していても、cblF型を否定することはできません。疾患の本態が「ビタミンの絶対的欠乏」ではなく「細胞レベルでの局所的な利用障害」であるためです。血清B12値が正常であることのみをもってcblF型を除外すると、診断の遅延につながります。
新生児マススクリーニング(NBS)
タンデム質量分析(MS/MS)を用いた新生児マススクリーニング(NBS)の普及により、無症状の段階で発見されるケースも増加しています。
💡 用語解説:タンデム質量分析法(MS/MS)
血液中の微量物質をその質量から同定・定量する分析装置を2台直列に接続した高感度・高特異的分析法です。新生児の濾紙血液から多種のアシルカルニチン・アミノ酸を同時測定でき、代謝異常症の早期発見に威力を発揮します。cblF型ではプロピオニルカルニチン(C3)の上昇・C3/C2比やC3/C0比の上昇・メチオニン(Met)の低下がスクリーニング指標となります。
遺伝子検査による確定診断
確定診断にはLMBRD1遺伝子の特異的な塩基配列解析が不可欠です。生化学的マーカーの異常パターンと組み合わせて、以下の遺伝子検査が選択されます。
🔬 コバラミン・ホモシステイン・メチオニン遺伝子検査
コバラミン代謝経路に関与する主要遺伝子を対象とした専門パネル。MMA・tHcy両方の上昇があり、cblF型が疑われる場合に有用。
🔬 メチルマロン酸尿症・ホモシスチン尿症NGSパネル
LMBRD1を含む複数のcbl型原因遺伝子を網羅したNGSパネル検査。複合ヘテロ接合変異の検出にも対応。
🔬 核・ミトコンドリアNGS遺伝子検査
核ゲノム・ミトコンドリアゲノムを包括的に解析する広域パネル。原因遺伝子が絞り込めていない代謝疾患の精査に有用。
7. 治療・長期管理と将来展望
cblF型異常症の現在の医学的マネジメントの目標は、不足している活性型補酵素(AdoCblおよびMeCbl)を外部から補充し、有毒な代謝産物(MMAおよびtHcy)の蓄積を最小化することにあります。脳神経系への不可逆的なダメージが蓄積する前のできる限り早期からの強力な介入が、患者の長期的なQOL(生活の質)と予後を決定づけます。
| 治療モダリティ | 具体的な介入内容 | 治療の目的・期待効果 |
|---|---|---|
| ヒドロキソコバラミン(OHCbl)大量補充 | 筋注(IM)または静注(IV)。初期:1〜10mg/日×3〜5日。維持:週数回〜月1回(代謝状態に応じる) | 治療の根幹。大量OHCblを血中に投与し、欠損リソソーム経路を部分的に迂回して非特異的経路で細胞質へB12を到達させる。シアノコバラミンより細胞内利用効率が高く推奨される。 |
| ベタイン(Betaine) | 50〜100 mg/kg/日(経口) | 肝臓の代替経路(ベタイン・ホモシステインメチルトランスフェラーゼ経路)を活性化してホモシステインをメチオニンに変換。tHcy毒性軽減と必須アミノ酸確保を同時に実現。 |
| L-カルニチン | 50〜100 mg/kg/日(経口) | 蓄積した有機酸(プロピオニル基など)と結合してカルニチンエステルを形成し、尿中排泄を促進。二次的カルニチン欠乏の予防にも必須。 |
| 葉酸(またはフォリン酸) | 5〜10 mg/日(経口) | メチオニン合成酵素機能低下による葉酸サイクルの停滞を支援。DNA合成障害(巨赤芽球性貧血)の改善に寄与。 |
| 栄養療法(食事管理) | 低タンパク質食・特殊フォーミュラの使用。長時間絶食の回避。 | MMAの前駆体となる特定アミノ酸(Ile・Val・Thr・Met)の過剰摂取を防ぎ代謝負荷を軽減。異化(カタボリズム)を防ぐ。 |
将来展望:遺伝子治療の可能性
現在の治療は極めて有効ですが、あくまで対症療法であり、生涯にわたる頻回な筋肉内注射と厳格な食事・代謝管理が必要なため患者・家族の負担は甚大です。cblF型患者から採取・培養された皮膚線維芽細胞に正常なLMBD1野生型cDNAを遺伝子導入した基礎研究において、細胞内でのコバラミン補酵素合成能と代謝機能が完全に回復することが生体外(in vitro)で実証されています。これはcblF型の細胞病態が、正常なLMBD1タンパク質を外来から補うことで完全可逆であることを意味します。アデノ随伴ウイルス(AAV)やCRISPR/Cas9ゲノム編集を用いたin vivo送達技術の確立により、近い将来に根治的な遺伝子治療が実現する可能性は極めて高いと考えられます。
8. 遺伝カウンセリングの意義
cblF型は常染色体潜性遺伝疾患であり、確定診断後には患者本人・兄弟姉妹・親族に対する丁寧な遺伝カウンセリングが不可欠です。
- ➤再発リスクの説明:両親がともに保因者の場合、次子が発症する確率は理論上25%です。保因者(罹患アレルを1本持つ)である確率は50%、非保因者は25%です。
- ➤保因者検査(キャリア検査):LMBRD1変異が同定されている場合、両親・兄弟姉妹に対して保因者検査を実施できます。特にc.1056delGという頻度の高い変異が存在するため、対象者への告知と検査の提案が重要です。キャリアスクリーニングとは何かについて詳しく解説しています。
- ➤出生前診断の選択肢:次子を希望する場合、既知の変異が同定されていれば羊水検査・絨毛検査による出生前遺伝子診断が可能です。また受精卵段階での着床前遺伝子検査(PGT-M)も選択肢の一つです。米国人類遺伝学会(ACMG)・ACOG推奨内容についても参考にしてください。
- ➤カップル(ブライダル)キャリアスクリーニング:結婚・妊娠前に保因者かどうかを調べるキャリアスクリーニングは、cblF型のような常染色体潜性疾患のリスクを妊娠前に把握する有効な手段です。
遺伝性疾患の診断・保因者検査・出生前診断は、患者・家族それぞれの人生観・価値観・家族計画と深く関わります。他の遺伝性疾患の患者ご家族の体験談として、副腎白質ジストロフィー保因者検査を受けた方の体験談や、ALD(副腎白質ジストロフィー)と家族計画もご参照ください。遺伝性疾患を持つ家族の意思決定プロセスに寄り添う姿勢が、臨床遺伝専門医によるカウンセリングの核心です。
よくある質問(FAQ)
🏥 コバラミン代謝異常・遺伝子検査についてのご相談
cblF型をはじめとするコバラミン代謝障害・先天性代謝異常症に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
参考文献
- [1] NCBI Gene. LMBRD1 gene (Gene ID: 55788). [NCBI Gene]
- [2] MedlinePlus Genetics. LMBRD1 gene. [MedlinePlus]
- [3] Orphanet. Methylmalonic aciduria and homocystinuria type cblF (ORPHA:79284). [Orphanet]
- [4] Rutsch F, et al. Identification of SLC19A3 as a novel transporter responsible for thiamine transport. Am J Hum Genet. 2011;88(1):5-17. LMBRD1: The gene for the cblF defect of vitamin B₁₂ metabolism. [PubMed 20446115]
- [5] Miousse IR, et al. Clinical and molecular heterogeneity in patients with the cblF inborn error of vitamin B12 metabolism. J Pediatr. 2011;158:335-341. [PubMed]
- [6] Deme JC, et al. Purification and interaction analyses of two human lysosomal vitamin B12 transporters: LMBD1 and ABCD4. Mol Membr Biol. 2014;31(7-8):250-261. [PubMed 25535791]
- [7] Xu X, et al. Cryo-EM structure of human lysosomal cobalamin exporter ABCD4. Nat Commun. 2022. [Semantic Scholar]
- [8] Bhatt DK, et al. LMBD1 protein serves as a specific adaptor for insulin receptor internalization. J Biol Chem. 2013;288(45):32424-32436. [PMC3820877]
- [9] Wang X, et al. Novel Nuclear Export Signal-Interacting Protein, NESI, critical for the assembly of hepatitis delta virus. J Virol. 2005;79(14):8889-8897. [PMC1143724]
- [10] Lmbrd1 expression is essential for the initiation of gastrulation. Mol Reprod Dev. 2016. [PMC4956942]
- [11] Phenotype puzzle: the role of novel LMBRD1 gene variant in Cbl deficiency causing Dyskeratosis Congenita-like clinical manifestations. Orphanet J Rare Dis. 2025. [PubMed 39939801]
- [12] Novel Parkinson’s Disease Genetic Risk Factors Within and Across European Populations (GP2, 2025). [PMC11957085]
- [13] Investigating the Genetic Relationship Between Vitamin B12 Metabolism and Parkinson Disease. Neurology Genetics. 2024. [Neurology Genetics]
- [14] GeneReviews: Disorders of Intracellular Cobalamin Metabolism. NCBI Bookshelf. [NBK1328]
- [15] OMIM #277380. Methylmalonic aciduria and homocystinuria, cblF type. [OMIM]






