目次

発達性てんかん性脳症1型(DEE1, OMIM:308350)は、X染色体短腕(Xp21.3)に位置するARX遺伝子の変異によって引き起こされるX連鎖性の重篤な神経疾患です。乳児期早期に難治性の点頭てんかんやサプレッション・バーストを呈する大田原症候群として発症し、不可逆的な精神運動発達の退行を伴います。本疾患の本質は、抑制性GABA作動性介在ニューロンの遊走障害に起因する「介在ニューロン病(Interneuronopathy)」であり、近年の分子メカニズム解明とケトン食療法(KDT)の有効性確立により治療パラダイムが大きく変化しています。
Q. DEE1(発達性てんかん性脳症1型)とはどのような病気ですか?
A. ARX遺伝子の変異により、生後1年以内に難治性のてんかん発作(点頭てんかん・大田原症候群)を発症するX連鎖性の重篤な神経発達疾患です。男児に多く、サプレッション・バーストやヒプスアリスミアと呼ばれる特徴的な脳波異常、重度の発達退行と知的発達症、しばしば全身性のジストニアを伴います。早期診断と適切な治療シーケンスの選択が予後を大きく左右します。
- ➤疾患の定義 → OMIM 308350、X連鎖性、生後1年以内発症の難治性てんかん性脳症
- ➤分子メカニズム → ARX→LMO1→CXCR4軸の破綻による介在ニューロン遊走障害
- ➤主な症状 → 大田原症候群→ウエスト症候群→レノックス・ガストー症候群への移行
- ➤鑑別診断 → XLAG・プラウド症候群・パーティントン症候群とのスペクトラム関係
- ➤治療戦略 → ACTH・ビガバトリン・ケトン食療法の最新エビデンス
1. DEE1とは:疾患の定義と症候群スペクトラム
発達性てんかん性脳症1型(Developmental and Epileptic Encephalopathy 1, DEE1)は、OMIM登録番号308350として国際的に認知された疾患で、生後1年以内(典型的には乳児期早期)に発症する重篤なてんかん症候群です。X染色体短腕(Xp21.3)に位置するARX(Aristaless-related homeobox)遺伝子の変異が直接的な原因となります。
💡 用語解説:発達性てんかん性脳症(DEE)とは
頻発するてんかん発作と、それに伴う重篤なてんかん性異常脳波が、発達途上の脳ネットワーク形成を直接的に阻害することで、進行性の認知・運動機能の退行や重度の知的発達症を引き起こす神経発達疾患群の総称です。「初期の神経発生異常」と「持続するてんかん性放電」が相互に悪影響を及ぼしながら独立して進行する点が、単純なてんかんとは異なる特徴です。次世代シーケンサーの普及により、DEEの約30〜50%が単一遺伝子の変異に起因することが明らかになっています。
DEE1は「同一の遺伝的基盤を持つ症候群スペクトラム」
DEE1は歴史的・臨床的文脈において、複数の名称で診断されてきました。これらは独立した疾患ではなく、同一のARX遺伝子変異を基盤とする一連の臨床的連続体(スペクトラム)として理解されています。
- ➤X連鎖性点頭てんかん症候群(ISSX1):男児に多発する点頭てんかんの遺伝形式を強調した名称
- ➤ウエスト症候群(点頭てんかん):乳児スパスム+ヒプスアリスミア+発達退行を3徴とする古典的症候群
- ➤大田原症候群(早期乳児てんかん性脳症):新生児期からのサプレッション・バーストを伴う最重症型
これらの症候群は患者の年齢や脳の発達段階に応じて互いに移行することが知られており、たとえば大田原症候群として発症した患者の約75%は、その後ヒプスアリスミアの出現を伴い点頭てんかん(ウエスト症候群)へと進展します。日本では大田原症候群が「指定難病146」として認定されています。
📌 DEE1の基本データ:OMIM 308350/責任遺伝子 ARX(Xp21.3)/遺伝形式 X連鎖性/発症時期 生後1年以内/罹患者は男児がほぼすべて/女性ヘテロ接合体の一部はX染色体不活化の偏りにより重症化することあり
2. ARX遺伝子と分子病態:介在ニューロン病という本質
ARX遺伝子は、胚発生における身体の多様な構造形成を制御するホメオボックス遺伝子ファミリーに属する重要な発生制御遺伝子です。ARXタンパク質は、高度に保存されたDNA結合領域である「ホメオドメイン」、C末端の「アリストアレスドメイン」、保存された「オクタペプチド」、そしてアラニン残基が連続して反復する「ポリアラニントラクト」を4箇所有しています。
💡 用語解説:転写因子(てんしゃいんし)とは
DNAの特定の配列に結合し、ほかの遺伝子の「読まれ方(発現)」を活性化したり抑えたりするタンパク質の総称です。ARXは中枢神経系において、ほかの遺伝子の発現を抑える「転写リプレッサー(抑制因子)」として機能します。発生段階の脳では脳室下帯などの増殖前駆細胞だけでなく、移動中および成熟後のGABA作動性介在ニューロンにおいて特異的に発現し続け、脳の正常なネットワーク構築を司ります。
「介在ニューロン病(Interneuronopathy)」というパラダイム
DEE1の根源的な病態は、大脳皮質における興奮性ニューロンと抑制性(GABA作動性)介在ニューロンの精緻なネットワークバランスの崩壊に帰着します。この抑制性ニューロンの機能不全あるいは数的な絶対的不足を指して、本疾患群は「介在ニューロン病(Interneuronopathy)」として再定義されています。
💡 用語解説:GABA作動性介在ニューロンとは
脳内でほかのニューロンの興奮を「抑える」役割を担う神経細胞の総称で、抑制性神経伝達物質GABA(γ-アミノ酪酸)を放出します。脳の発生初期には腹側終脳の神経節隆起(Ganglionic Eminence: GE)で誕生し、そこから接線方向に長距離を移動して大脳皮質に到達します。脳全体のニューロンの10〜15%を占めるに過ぎませんが、興奮と抑制のバランス(E/Iバランス)を保ち、てんかん発作を防ぐうえで決定的に重要です。
分子メカニズム:ARX → LMO1 → CXCR4 の脱抑制カスケード
最新の単一細胞RNAシーケンス(scRNA-seq)とクロマチン免疫沈降シーケンス(ChIP-seq)を駆使した研究により、ARXが介在ニューロンの遊走を制御する分子メカニズムが解明されました。鍵となるのは「ARX→LMO1→CXCR4」という二重抑制カスケードです。
ARX遺伝子によるGABA作動性介在ニューロン遊走制御の分子カスケード
介在ニューロンの誕生地
ARXがLMO1を抑制 → LMO1がCXCR4を抑制できなくなる
→ CXCR4が正常に発現 → ガイダンス受容体として機能
介在ニューロンの正常な定着
ARXは転写リプレッサーとしてLMO1を抑制する。LMO1は通常、皮質ガイダンス受容体CXCR4を抑制するため、ARXがLMO1を抑える=結果的にCXCR4が発現できる、という二重抑制(脱抑制)の仕組み。ARXが変異により機能を失うとLMO1が過剰となり、CXCR4が低下し、介在ニューロンは目的地を見失う。
この論理的な抑制カスケードを実際の脳の発生過程に重ねると、神経節隆起(MGE/CGE)で誕生した介在ニューロンが大脳皮質へと遊走していくダイナミックな細胞移動として可視化できます。
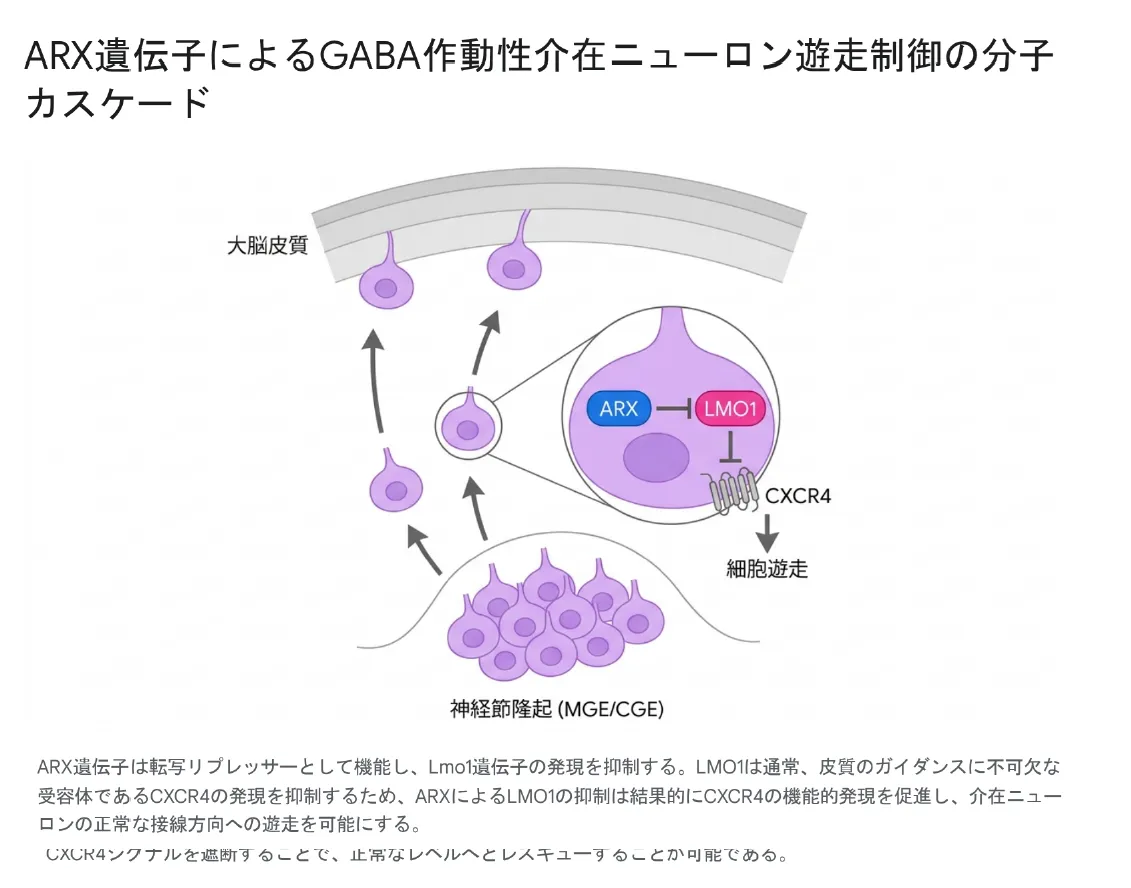
ARX遺伝子は転写リプレッサーとして機能し、Lmo1遺伝子の発現を抑制する。LMO1は通常、皮質のガイダンスに不可欠な受容体であるCXCR4の発現を抑制するため、ARXによるLMO1の抑制は結果的にCXCR4の機能的発現を促進し、介在ニューロンの正常な接線方向への遊走を可能にする。
💡 用語解説:ポリアラニン反復伸長変異
ARXタンパク質には、アミノ酸の一種である「アラニン」が連続して並ぶ4つのポリアラニントラクトがあります。DEE1で最も頻度が高い変異は、第1または第2ポリアラニントラクトの異常な伸長です。たとえば第1トラクトに33塩基対の重複が生じると、本来16個のアラニン残基が27個にまで拡張されます。GCG繰り返し配列が「不等交差」や「複製スリッページ」を起こしやすいゲノム構造上の脆弱性が背景にあります。タンパク質を完全に無効化するわけではなく機能を部分的に低下させるため、巨視的な脳構造を保ちつつ介在ニューロンの遊走障害を引き起こします。
3. 主な症状と脳波の経時的進展
DEE1の臨床像の最大の特徴は、生後1年以内(多くは生後数ヶ月以内)に発症する難治性のてんかん発作と、それに伴う壊滅的な精神運動発達の退行です。日本における代表的な報告例では、生後1ヶ月から上肢の屈曲を伴う強直スパスムが観察され、生後3ヶ月で不随意運動とともに専門機関を受診し、視覚的反応や定頸(首のすわり)が認められず、全体的な筋緊張低下を呈する経過が記載されています。
発作型と脳波パターンは年齢とともに変遷する
DEE1の患者は、成長と脳の成熟に伴って発作の形態と脳波パターンが特徴的な変遷をたどり、それに呼応して臨床診断名(症候群名)が移行することが多いのが特徴です。
🩺 生後早期(0〜3ヶ月)
大田原症候群
発作型:強直スパズム、部分発作
脳波:サプレッション・バーストパターン(高振幅のバーストとほぼ平坦な抑制期が数秒間隔で交互に出現)
🩺 乳児期(3ヶ月〜1歳)
ウエスト症候群
発作型:乳児スパスム(体幹を前屈・四肢硬直のジャックナイフ様発作)が数分にわたり群発
脳波:ヒプスアリスミア(無秩序で極めて高振幅の徐波と棘波が混在するカオス的パターン)
🩺 幼児期以降(1〜5歳〜)
レノックス・ガストー症候群
発作型:非定型欠神発作、強直発作、脱力(アトニー)発作、間代発作などが混在
脳波:汎発性遅棘徐波、多棘徐波。ヒプスアリスミアは消失
💡 用語解説:サプレッション・バーストとヒプスアリスミア
サプレッション・バーストとは、脳波上で「高電圧の鋭い棘波・徐波(バースト)」と「ほぼ平坦な抑制期(サプレッション)」が数秒〜十数秒間隔で交互に現れるパターンです。新生児期の大田原症候群を強く示唆します。
ヒプスアリスミアは、全体的に無秩序で極めて高振幅の徐波と棘波が混在するカオス的・無構造な脳波パターンで、ウエスト症候群(点頭てんかん)の中核的所見です。これらのてんかん性放電そのものが脳の正常なシナプス形成を物理的・電気的に阻害するため、発作の発症と同時に乳児はそれまで獲得していた寝返りや喃語といった発達マイルストーンを喪失していきます。
てんかんとは独立した「重篤なジストニア」の合併
ARX関連DEE1の特筆すべき臨床的特徴として、てんかんとは独立した重篤な運動障害(ジストニア)の合併が挙げられます。第1ポリアラニントラクトの伸長変異を持つ男児の追跡調査では、生後2〜3ヶ月で点頭てんかんを発症した後、生後6ヶ月頃から全身性のジストニアが出現し、2歳までに頭部のコントロール不良、体幹の筋緊張低下、そして重度のジストニア性四肢麻痺へと進行する症例が複数報告されています。
中には、生命を脅かす「ジストニア重積状態(status dystonicus)」を反復する例も報告されており、脳MRIでは被殻に小空洞化やT2強調画像での高信号領域など、進行性の組織喪失を示唆する所見が認められることがあります。周産期仮死などの明確な病歴がないにもかかわらず、乳児スパスムとジスキネジア性脳性麻痺を合併する男児においては、ARX遺伝子の変異を第一に疑うことが診断の鍵となります。

4. 鑑別診断:ARX関連疾患スペクトラム
🔍 関連記事:XLAG(X連鎖性滑脳症)|プラウド症候群|パーティントン症候群|MRX29|DEE8
ARX遺伝子の変異は単一の疾患にとどまらず、ほぼ連続的な表現型スペクトラムを形成します。致死的な脳形成異常を伴う重篤な疾患から、脳の構造的異常を伴わない比較的軽度の知的発達症まで多岐にわたります。100種類以上の病原性バリアントが報告されており、変異の性質(ミスセンス、ナンセンス、ポリアラニン伸長など)とタンパク質内の位置が、臨床的重症度を決定づける極めて強力な予測因子となります。
💡 用語解説:機能喪失と部分的機能低下の違い
XLAGに見られるような重篤な脳形成異常は、ARXタンパク質の完全な機能喪失(loss of function)によって引き起こされます。これにより神経前駆細胞の増殖自体が根本から阻害され、大脳皮質の形成が停止します。一方、DEE1で多いポリアラニン伸長は、タンパク質を完全に無効化するわけではなく細胞内発現量の減少や半減期の変化を介して機能を部分的に低下させます。この「不完全な機能低下」こそが、巨視的な脳構造を維持しつつも介在ニューロンの局所的な遊走障害とてんかん原性の獲得という微細な病態を引き起こす原因です。
5. 診断と遺伝子検査の進め方
DEE1を疑うべき臨床的レッドフラッグ
💡 ARX関連DEE1を強く疑う臨床所見
- ➤男児での生後1年以内発症の難治性てんかん(特に強直スパズムや乳児スパスム)
- ➤サプレッション・バーストまたはヒプスアリスミアの脳波所見
- ➤てんかんと並行して進行する全身性ジストニア
- ➤周産期歴に問題がないにもかかわらずジスキネジア性脳性麻痺様の運動障害
- ➤家族歴にX連鎖性知的発達症・てんかん男児が複数いる場合
分子遺伝学的検査:NGSパネル検査が標準アプローチ
DEE1の確定診断にはARX遺伝子の塩基配列解析が必要です。ただし新生児期・乳児期発症のてんかん性脳症は責任遺伝子が極めて多岐にわたるため、単一遺伝子検査ではなく、関連する数百の遺伝子を一度に解析するNGS(次世代シーケンサー)パネル検査が推奨されます。ポリアラニン反復伸長の検出にはサンガー法による配列確認や、必要に応じて反復長を測定する追加解析が有用です。
💡 用語解説:NGSパネル検査とは
次世代シーケンサー(Next Generation Sequencing)を用いて、特定の疾患グループに関連する複数の遺伝子を一度に解析する検査です。新生児てんかんに対しては約285遺伝子、思春期・成人発症を含むてんかんでは1057遺伝子といった大規模パネルが利用可能で、原因不明のてんかんに対する診断率を従来のサンガー法から大きく向上させています。一回の血液採取で広範な解析ができ、結果として治療選択(ビタミンB6依存性てんかんへのB6投与、グルコーストランスポーター1欠損症へのケトン食療法など)に直結する有用な情報が得られます。
脳画像検査の役割:スペクトラム上の位置づけ
DEE1単独では脳MRI上の巨視的な形成異常は基本的に認められませんが、軽度の脳梁低形成や小頭症を伴うことがあります。一方、滑脳症や脳梁完全欠損が認められた場合はXLAGを強く疑います。被殻における進行性の組織喪失(小空洞化、T2強調画像での高信号領域)が認められれば、ジストニア重積を伴うDEE1の特徴的な画像所見である可能性があります。
6. 治療と長期管理:最新の治療シーケンス
DEE1を含む難治性の乳児てんかん性スパスム症候群(IESS)に対する治療の最終目標は、発作の完全な抑制と発達退行の阻止です。しかしARX変異によるてんかんは極めて難治性であり、フェノバルビタールやバルプロ酸などの従来の抗てんかん薬単独では効果が乏しいのが実情です。
第一選択薬:ACTH療法とビガバトリン
💉 ACTH療法
副腎皮質刺激ホルモンの筋肉内注射。コルチゾール分泌の促進と脳内メラノコルチン受容体への直接作用により神経興奮を抑制。短期的には劇的な発作抑制をもたらすことが多いが、重度の免疫抑制・高血圧・電解質異常・心筋肥大などのリスクで継続困難。投薬終了後の再発も多い。
💊 ビガバトリン
GABA分解酵素(GABAトランスアミナーゼ)を不可逆的に阻害し、シナプス間隙のGABA濃度を上昇させて抑制性伝達を増強。結節性硬化症に伴う点頭てんかんに特に有効性が高い。網膜毒性による不可逆的視野狭窄のリスクがあり、定期的な眼科的モニタリングが必須。
これらの第一選択治療を用いても、患者の25〜50%は初期治療に反応しない(不応性)か、あるいは早期に再発をきたすのが実情です。そのため、不応例に対する次の一手をどう設計するかが治療成績を左右します。
ケトン食療法(KDT)の有効性:治療シーケンスの新標準
💡 用語解説:ケトン食療法(KDT)とは
高脂肪・極低炭水化物・適正タンパク質の食事を厳密に管理することで、体内のエネルギー代謝をグルコース依存からケトン体(アセト酢酸、β-ヒドロキシ酪酸)依存へと切り替える治療法です。ケトン体は脳血液関門を通過し、ミトコンドリアのエネルギー産生効率を向上させるとともに、神経細胞の過剰興奮を直接的およびエピジェネティックに抑制する機序を持つと考えられています。100年以上の使用実績があり、ドラベ症候群・点頭てんかん・薬剤抵抗性てんかんなど幅広い適応があります。
最新の比較研究において、IESS患者に対するKDTとACTHの逐次治療の成績が興味深い結果を示しています。
乳児てんかん性スパスム:治療順序別の累積奏効率
45%
19%
14%
KDT先行群
KDT→ACTH→ASM
累積 78%
18%
50%
ACTH先行群
ACTH→KDT→ASM
累積 72%
ポイント:ACTH不応後にKDTを導入した患者の50%が持続的な発作消失を示したのに対し、KDT不応後にACTHを導入して奏効したのは19%でした。最終的な累積奏効率は両群間でほぼ同等(78% vs 72%)に達しており、KDTが極めて有効な治療選択肢であることを示しています。
別のレトロスペクティブ調査でも、ACTHやビガバトリンに不応であった18名にKDTを導入した結果、導入2ヶ月後に39%の患者が発作頻度を50〜90%減少させ、44%が90%以上の顕著な減少を達成。この抗てんかん効果は6ヶ月後・12ヶ月後も持続し、嘔吐や便秘などの副作用が見られたのは17%にとどまりました。腎結石や骨粗鬆症などの重篤な有害事象は極めて稀です。
長期予後と多職種連携による合併症管理
かつてDEEは小児科学の領域に限定されて議論されてきましたが、近年は医療・栄養・呼吸管理の進歩により成人期に到達するDEE患者が増加し、新たな臨床的課題が浮き彫りになっています。大規模な死亡率コホート調査では、DEE患者の死亡の80%が「非SUDEP(てんかん突然死以外の原因)」であることが明らかになりました。
🫁 呼吸器・嚥下管理
嚥下障害に起因する誤嚥性肺炎の予防、慢性的な気道管理、必要に応じた胃瘻造設や気道評価。
🦴 整形・リハビリ
側弯症・関節拘縮の予防と治療、ジストニアに対するボツリヌス治療やバクロフェン療法。
🍽️ 栄養管理
体重・成長の評価、ケトン食療法を継続する場合の電解質・脂質管理、ビタミン・ミネラル補充。
🧠 神経変性への備え
成人期における進行性の脳萎縮や運動障害の悪化を念頭に置いた長期画像フォローと支援体制。
遺伝性DEEは初期の「神経発達障害」フェーズに続いて、加齢に伴う累積的な神経生物学的ストレスが引き起こす「進行性の神経変性」の軌跡をたどる二相性疾患であることが、近年の死後脳病理研究や神経画像研究から示唆されています。将来の治療介入は「単なる発作の抑制」から「神経保護と変性の阻止」へと拡張されつつあります。
7. 遺伝カウンセリング:女性キャリアと家族計画
🔍 関連記事:X染色体連鎖劣性遺伝|遺伝形式|キャリアスクリーニングとは|米国人類遺伝学会の推奨
X連鎖性遺伝の基本と再発リスク
DEE1はX染色体上のARX遺伝子変異に起因するX連鎖性疾患です。母親が変異アレルを保因している場合、その息子は50%の確率でDEE1を発症し、娘は50%の確率で保因者となります。一方、父親がARX変異を持つ症例は通常生殖年齢まで生存しないため、父親由来の遺伝はほとんど考慮されません。
💡 用語解説:X染色体連鎖劣性遺伝とは
原因遺伝子がX染色体上に存在し、変異が「劣性(潜性)」の形質として遺伝する形式です。男児(XY)はX染色体を1本しか持たないため、X染色体上の病的変化があると発症しやすい一方、女児(XX)は通常もう1本のXが補うため無症状の保因者になることが多いのが特徴です。ただし女性でも、後述する「X染色体不活性化(Lyonization)の偏り」により軽度〜中等度、まれに男児並みの症状が出ることがあります。
女性キャリアの臨床的多様性とX染色体不活化の偏り
通常、変異アレルを1つ持つ女性(ヘテロ接合体)は無症状か軽度です。発生初期の胚において2本のX染色体のうち1本がランダムに不活化される「X染色体不活化(XCI)」という遺伝子量補償機構が働くためです。しかし臨床現場では、男性患者に匹敵するほどの重度の知的発達症・難治性てんかん・脳梁欠損などを呈するARX変異の女性キャリアが少なからず報告されており、これは「X染色体不活化の偏り(Skewed X-inactivation)」によって説明されます。
💡 用語解説:X染色体不活化の偏り(Skewed X-inactivation)
通常はランダムに50:50で行われるはずのX染色体不活化が、何らかの理由で一方のX染色体に偏ってしまう現象です。一方のアレルが全細胞の75%以上で活性化されている状態を「偏り」、90%以上を「極端な偏り」と定義します。原因としては、(1) 胚着床期の細胞数が小さいタイミングでの偶発的な確率の偏り、(2) 変異X染色体が活性化している細胞の生存・増殖上の不利益による選択的淘汰、の2つが知られています。X連鎖性知的発達症の保因者の約50%で80:20以上の著しい偏りが観察されたという報告もあります。
重篤な症状を呈する女性キャリアに対しては、てんかんや発達遅滞の神経学的管理のみならず、心血管系のスクリーニング、精神的ケア、将来の家族計画に向けた遺伝カウンセリングを含む包括的な医療アプローチが推奨されます。同じ家系の女性であっても症状の出方が大きく異なるため、ケースごとの丁寧な評価が欠かせません。
出生前診断と次子妊娠への備え
家系内のARX変異が同定されている場合、次子の妊娠に対して以下のような選択肢があります。いずれもメリット・デメリット、心理的・倫理的な問題が複雑に絡むため、遺伝カウンセリングの活用が必須です。
- ➤絨毛検査・羊水検査による出生前確定診断:家系内変異が分かっている場合、胎児の遺伝子型を確実に評価できます。胎児が男児で変異を持つ場合の対応について、ご夫婦であらかじめ十分な話し合いと心理的準備が必要です。
- ➤着床前遺伝学的検査(PGT-M):体外受精と組み合わせて、移植前に変異を持たない胚を選別する選択肢。施設・倫理委員会の承認が必要です。
- ➤非侵襲的検査の可能性:性別判定を含むNIPTは、X連鎖性疾患の家系で胎児が女児であることが判明すれば家族の不安を軽減できる場合があります。ただし変異そのものの検出には絨毛・羊水検査が必要です。
参考までに、別のX連鎖性疾患である副腎白質ジストロフィー(ALD)の保因者検査の体験談や、ALDと家族計画の選択肢についての記事は、X連鎖性遺伝病の保因者の意思決定プロセスの参考になります。
8. よくある誤解
誤解①「点頭てんかんは原因不明が多い」
かつては「症候性」「特発性」「潜因性」に分類されていましたが、現在は遺伝子検査で原因が特定できるケースが大幅に増えています。ARXは男児の点頭てんかんの主要な遺伝的原因の1つであり、早期の遺伝子検査は治療選択を変えうる重要な情報をもたらします。
誤解②「ACTHが効けばOK、再発したら諦める」
ACTH不応・再発例に対するケトン食療法の有効性は最新研究で明確に裏付けられています。ACTH不応後にKDTを導入した患者の50%が持続的な発作消失を示しました。早期のKDT導入を選択肢として検討する価値があります。
誤解③「DEE1は静的な脳症」
かつてDEEの障害は乳児期に固定された静的状態と考えられていましたが、成人期に達した患者の死後脳研究や神経画像研究から、加齢に伴う進行性の神経変性が起こる二相性疾患であることが示されています。長期的なフォローと神経保護的アプローチが新しい課題です。
誤解④「女性の保因者は症状が出ない」
通常は無症状ですが、X染色体不活化の偏りにより男児並みの重症化を呈する女性キャリアが報告されています。「無症状」を前提にせず、症状の有無にかかわらず保因者には包括的な医療フォローが必要です。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 難治性てんかん・遺伝性疾患の診断・遺伝カウンセリング
DEE1をはじめとする遺伝性てんかん性脳症に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] OMIM #308350. Developmental and Epileptic Encephalopathy 1; DEE1. Johns Hopkins University. [OMIM]
- [2] MedlinePlus. Developmental and epileptic encephalopathy 1. National Library of Medicine. [MedlinePlus]
- [3] MedlinePlus. ARX gene. National Library of Medicine. [MedlinePlus]
- [4] Friocourt G, Parnavelas JG. Mutations in ARX Result in Several Defects Involving GABAergic Neurons. Front Cell Neurosci. 2010. [PMC2841486]
- [5] ARX mutation-associated interneuron defects provide insights into mechanisms underlying developmental epilepsies. Brain. 2025. [Oxford Academic]
- [6] Colombo E, et al. Arx Is a Direct Target of Dlx2 and Thereby Contributes to the Tangential Migration of GABAergic Interneurons. J Neurosci. 2008;28(42):10674-10686. [J Neurosci]
- [7] Guerrini R, et al. Expansion of the first PolyA tract of ARX causes infantile spasms and status dystonicus. Neurology. 2007. [Neurology]
- [8] Absoud M, et al. A Longer Polyalanine Expansion Mutation in the ARX Gene Causes Early Infantile Epileptic Encephalopathy with Suppression-Burst Pattern (Ohtahara Syndrome). Am J Hum Genet. 2007. [PMC1950814]
- [9] Olivetti PR, Noebels JL. Interneuron, Interrupted: Molecular Pathogenesis of ARX Mutations and X-linked Infantile Spasms. Curr Opin Neurobiol. 2012. [PMC3437236]
- [10] Skewed X-Chromosome Inactivation Is a Common Feature of X-Linked Mental Retardation Disorders. Am J Hum Genet. [PMC384975]
- [11] Role of the Ketogenic Diet Therapy and ACTH as Second Treatments in Drug-Resistant Infantile Epileptic Spasms Syndrome. Nutrients. 2025;17(13):2085. [MDPI]
- [12] Progress of ketogenic diet in the treatment of developmental epileptic encephalopathy. Front Pediatr. 2025. [PMC12358386]
- [13] Adults with Developmental and Epileptic Encephalopathies in the Precision Treatment Era. NeurologyLive. [NeurologyLive]
- [14] 大田原症候群(指定難病146). 難病情報センター. [難病情報センター]
- [15] Type 1 early infantile epileptic encephalopathy: A case report. Medicine. 2024. [PMC10891437]






