目次

ARX遺伝子は、脳の中で「ブレーキ役」をはたく抑制性神経細胞(GABA作動性ニューロン)の発生・遊走・成熟を司るマスターレギュレーターです。X染色体上にあり、変異が起こると大田原症候群・West症候群・X連鎖性滑脳症(XLAG)など、極めて多彩な神経疾患を引き起こします。一つの遺伝子から驚くほど幅広い表現型が生まれる「ARXopathies」の分子基盤と、近年急速に進展する治療開発まで、臨床遺伝専門医の視点で解説します。
Q. ARX遺伝子はどんな働きをしている遺伝子ですか?
A. 脳の発生過程で、抑制性神経細胞(GABA作動性ニューロン)が「正しい場所」へ「正しいタイミング」で移動できるよう指揮する転写因子をつくる遺伝子です。X染色体上にあり、ホメオドメインを介して多数の標的遺伝子の発現を制御します。変異により致死的な滑脳症から、難治性てんかん、知的障害単独まで極めて広いスペクトラムの疾患を引き起こします。
- ➤遺伝子の基本情報 → ヒトX染色体Xp21.3、5エキソン、562アミノ酸の転写因子
- ➤主要ドメイン → ホメオドメイン・4つのポリアラニントラクト・Engrailedドメイン
- ➤関連疾患 → XLAG・大田原症候群・West症候群・Partington症候群・Proud症候群など
- ➤最頻変異 → c.428_451dup24(第2ポリアラニントラクト伸長)が脳形成異常なし症例の45〜76%
- ➤最新治療 → AMD3100・ASO・AAV遺伝子治療・抑制性神経細胞移植(NRTX-1001)
1. ARX遺伝子の基本情報
ARX(Aristaless Related Homeobox)遺伝子は、ヒトX染色体短腕の Xp21.3 領域に位置するX連鎖性遺伝子です。ゲノム上で約11kb(キロベース)の領域を占め、5つのエキソンとそれを結ぶイントロンから構成されています。転写されたmRNAは1,686塩基対のオープンリーディングフレームを持ち、562個のアミノ酸からなる転写因子タンパク質をコードしています。
💡 用語解説:転写因子とは
DNAの遺伝情報からRNAをつくる「転写」というプロセスを制御するタンパク質のことです。遺伝子の「スイッチ」を入れたり切ったりする役割を持ちます。ARXタンパク質は、特定のDNA配列に結合して下流の遺伝子の発現を強力に「抑制」する転写抑制因子として主に働きます。一つの転写因子が数百〜数千の標的遺伝子を制御できるため、ARXのような転写因子の異常は広範な発生プロセスに同時に影響を与えるのです。
ARXタンパク質は、ショウジョウバエの aristaless(al)遺伝子から進化的に保存されてきた脊椎動物オルソログであり、グループII aristaless関連タンパク質ファミリーに分類されます。発生過程の特定のタイミングで強く発現し、神経幹細胞の増殖維持と、抑制性神経細胞の運命決定・遊走・成熟という、脳の構築にとって最も基本的なプロセスを統括しています。
💡 用語解説:抑制性神経細胞(GABA作動性ニューロン)とは
脳の中には大きく分けて「興奮性」と「抑制性」の2種類の神経細胞があります。興奮性ニューロンが「アクセル」だとすれば、抑制性ニューロン(GABA作動性ニューロン)は「ブレーキ」の役割を担います。GABA(γ-アミノ酪酸)という神経伝達物質を使って、過剰な興奮を抑え、神経回路のバランスを整える働きを持ちます。このブレーキが効かなくなると、てんかん発作や認知機能障害が引き起こされます。ARX遺伝子は、まさにこの「ブレーキ役」のニューロンの誕生と配置を司る指揮者なのです。
ARXは中枢神経系だけでなく、膵臓・精巣・骨格筋の発生にも関与しており、これがARX変異患者で外性器異常や慢性下痢などの全身症状が見られる理由となっています。X連鎖性ホメオボックス遺伝子の一つとして、prd様クラスホメオボックス遺伝子ファミリーに属しています。
2. ARXタンパク質の構造とドメイン
ARXタンパク質が標的遺伝子の転写を精密に制御できる理由は、その内部に配置された4つの主要な機能ドメインと、それぞれが結合する補助因子(コファクター)にあります。各ドメインの違いがそのまま、変異の種類による表現型の違いを生み出します。
🧬 ホメオドメイン
prd様(paired-like)クラスに属する60アミノ酸のDNA結合領域。標的遺伝子の特定の塩基配列を認識して結合する「鍵」の部分です。ここに変異が起こるとDNA結合能が完全に失われ、最重症のXLAG(X連鎖性滑脳症)が引き起こされます。
🔁 4つのポリアラニントラクト
アラニンというアミノ酸が連続して並ぶ領域が4箇所あります。第1・第2トラクトの伸長変異が、ARX関連疾患全体の約59%を占める最頻変異タイプです。
🔇 Engrailed/Octapeptideドメイン
転写抑制活性の中核を担うドメイン。Transducin-like enhancer(TLE)タンパク質を補助因子としてリクルートし、下流遺伝子の発現を強力にオフにします。
📍 C末端Aristalessドメイン
タンパク質のC末端側に位置する保存領域。タンパク質間相互作用や転写活性の空間的調節に関わります。OARドメインとも呼ばれます。
💡 用語解説:ホメオドメインとは
60アミノ酸からなり、「ヘリックス・ターン・ヘリックス」と呼ばれる立体構造でDNAに結合するタンパク質ドメインです。ホメオボックス遺伝子がコードしており、生物の発生における体の前後・背腹のパターン形成を司る最も基本的な転写因子グループです。ヒトには約230個のホメオドメインタンパク質が存在し、ARXもその一つです。ARXのホメオドメインに変異が入ると、DNAという「楽譜」を読めなくなった指揮者のように、下流遺伝子の発現制御が完全に破綻します。
💡 用語解説:ポリアラニントラクトとは
タンパク質を構成するアミノ酸のうち、「アラニン」という小さなアミノ酸が連続して並んだ領域のことです。ARXには通常、第1トラクト16個・第2トラクト12個などの長さで4つのトラクトが存在します。これがDNA上の「GCG」配列の重複によって異常に伸長すると、タンパク質の折りたたみが乱れ、機能が変化したり、細胞内で凝集塊(インクルージョン)を形成したりします。「ポリアラニン伸長病」は、ハンチントン病で有名なポリグルタミン病とは異なる独自の疾患カテゴリーを形成しています。
3. 脳発生におけるARXの役割
大脳皮質という極めて複雑な構造は、興奮性の錐体細胞と抑制性の介在神経細胞(インターニューロン)の精緻なバランスの上に成り立っています。ARXはこのうち、後者の介在神経細胞の旅路を最初から最後まで導く役割を担います。
介在神経細胞の長距離「接線方向遊走」を統括する
大脳皮質の介在神経細胞は、胎生期にお腹側の「神経節隆起(GE)」という場所で誕生します。そこから大脳皮質という遠い目的地まで、長い距離を「接線方向(Tangential migration)」に移動するという、極めてユニークな旅をします。ARXはこの旅のいつ出発するか・どの方向に進むか・どこで停止するかを、複数の下流遺伝子を介して厳密にコントロールします。
🧬 ARXが直接制御する3つの主要シグナル経路
- ➤Nkx2.1の抑制:細胞が「出発」のタイミングを掴むための転写因子の制御
- ➤CXCR4-CXCL12軸の維持:大脳皮質に向かう「化学コンパス」の感度調整
- ➤Nrg1/ErbB4経路の制御:方向性を持った遊走を支える誘導シグナル
特に重要なのは、ARXがGABA作動性ニューロンの「親分」にあたる転写因子Dlx1/2の直接の下流に位置することです。Dlx1/2が「お前はGABA作動性ニューロンになれ」と運命を決めた後、ARXが「次は皮質まで歩いていくんだぞ」と移動の実行を担当する——という分業構造になっています。
神経幹細胞の増殖タイミングも制御
遊走中の介在神経細胞だけでなく、ARXは背側脳室帯(VZ)と脳室下帯(SVZ)に存在する神経前駆細胞の増殖と分化のタイミングも厳密に制御しています。細胞周期阻害因子(Cdkn1cなど)を抑制することで前駆細胞が早すぎる分化に進むのを防ぎ、「未分化な増殖プール」を適切なサイズで維持します。ARXが欠損するとこのバランスが崩れ、皮質の層構造形成に重大な影響が出ます。
嗅球と内分泌器官(膵臓・精巣)でも活躍
ARXは大脳皮質以外でも、嗅球のドーパミン作動性介在神経細胞の分化を誘導します(Nurr1の上流で機能)。さらに膵臓ではグルカゴンを産生するα細胞のアイデンティティを確立し、同時にインスリン産生β細胞への運命転換を抑制するという真逆の働きをします。精巣では生殖腺の正常な分化に関与しており、ARX変異男児で曖昧な外性器が見られる原因となっています。
4. ARX関連疾患(ARXopathies)のスペクトラム
ARX遺伝子にはこれまでに100家系以上から44種類を超える変異が報告されており、それらは少なくとも10種類の明確に定義された臨床的症候群を引き起こします。ARX変異の最大の特徴は、同一の変異を持つ家系内・兄弟間でも全く異なる重症度や症状を呈するという顕著な多面発現性(Pleiotropy)と遺伝学的異質性です。
これらの疾患群は、MRIで脳形成異常が確認できるかどうかで大きく2つのグループに分けられ、変異の分子的性質(機能喪失型か、機能変化型か)と強く相関します。
🧠 脳形成異常を伴う重症型
変異タイプ:タンパク質の機能を完全に失わせる変異(ナンセンス・フレームシフト・大規模欠失・ホメオドメイン変異)
- XLAG(X連鎖性滑脳症および外性器異常):周産期〜乳児期致死
- Proud症候群:脳梁欠損+外性器異常
- 水無脳症:大脳半球の広範な欠損
⚡ 脳形成異常を伴わない群
変異タイプ:機能が部分的に保たれるミスセンス変異・ポリアラニン伸長変異
- 大田原症候群(DEE1・早期乳児てんかん性脳症)
- West症候群(X連鎖性点頭てんかん・ISSX)
- Partington症候群:手指ジストニア+知的障害
- 非症候群性XLMR(MRX29):知的障害単独
💡 用語解説:滑脳症(かつのうしょう / Lissencephaly)とは
「滑らかな脳」を意味するギリシャ語に由来する先天性の脳形成異常です。健康な脳の表面には多数のひだ(脳回)と溝(脳溝)がありますが、滑脳症ではこれらが欠損または減少し、脳の表面が滑らかになっています。胎生期に神経細胞が正しい場所まで移動できないことが原因。XLAGはこの滑脳症の一型で、ARX遺伝子の機能喪失型変異が原因です。重度の知的障害・難治性てんかん・運動障害を引き起こします。当院では滑脳症NGSパネル検査で原因遺伝子を網羅的に調べることが可能です。
5. 変異タイプと表現型の対応
最頻変異 c.428_451dup24 と臨床的多面発現
全ARX変異の中で最も頻度が高く、脳形成異常を伴わない症例の45〜76%を占めるのが、エクソン2における24塩基対のインフレーム重複変異(c.428_451dup24)です。この変異は、第2ポリアラニントラクトを通常の12アラニンから20アラニンへと8つ伸長させます。
この単一変異がARX関連疾患の「多面発現性(Pleiotropy)」という概念を最も明確に示しています。同じ変異を持つ18名の男性患者を対象とした大規模家系調査では、症状の出現頻度が次のように極めて多彩であることが確認されました。
📊 c.428_451dup24変異を持つ18名の症状出現頻度
同一の遺伝子変異が、患者ごとに全く異なる症状の組み合わせを生み出す
同一遺伝子変異でも症状の組み合わせは患者ごとに異なる。未知の修飾遺伝子・エピジェネティック因子・環境要因が表現型を左右していると考えられる
分子メカニズムの違いが運命を分ける
同じ「機能喪失型変異」でも、興味深い例外が存在します。c.81C>G(p.Y27X)変異はわずか26アミノ酸でタンパク質を未成熟終結させるはずのナンセンス変異ですが、患者は致死的なXLAGではなく大田原症候群やWest症候群を発症しました。これは下流の開始コドン(位置41のメチオニン)からmRNA翻訳が「再開」され、N末端を欠いた部分機能性タンパク質が産生されるという、独特の救済機構が働くためと考えられています。
また、双子の患者で大田原症候群を引き起こしたエクソン5の単一アミノ酸置換 c.1604T>Aの例から、臨床的重症度はポリアラニン伸長の物理的長さだけでなく、変異がタンパク質全体の折りたたみや相互作用ネットワークに与える「質的な影響度」に強く依存することが明らかになっています。
6. 女性キャリアにおける表現型
ARXはX連鎖性遺伝子のため、男性(半接合体)で重篤な症状を呈するのが基本ですが、変異アレルをヘテロ接合で持つ女性キャリアにおいても無視できない頻度で症状が現れます。これはX連鎖性遺伝病における女性キャリアの一般的な特徴であり、丁寧な遺伝カウンセリングが不可欠です。
💡 用語解説:X染色体の不活化(X-inactivation)
女性は2本のX染色体を持ちますが、発生のごく初期に細胞ごとにランダムに「片方のX染色体だけがオフになる」現象が起こります。これにより、女性の体はモザイク状に「父由来のXが活動している細胞」と「母由来のXが活動している細胞」が混在した状態になります。ARXのような変異を持つ女性キャリアでは、たまたま正常Xがオフになった細胞の比率が偏る(偏向X不活化:Skewed X-inactivation)と、変異の影響が強く出てしまうことがあります。これが同じ家系内の母娘・姉妹で症状の重さが大きく違う理由です。
ARX変異を持つ女性キャリアで報告されている所見は極めて幅広いものです:
- ➤無症状〜軽度の学習障害:多くの女性キャリアはこの範囲
- ➤脳梁欠損(ACC):構造的な異常として最も特徴的
- ➤不安障害・うつ病:精神症状として一定頻度
- ➤難治性てんかん・知的障害:偏向X不活化が強い場合

7. 最新研究:ヒト脳オルガノイドが覆したパラダイム
ARX関連疾患の研究は、これまで遺伝子改変マウスモデルに大きく依存してきました。Kitamura P355R/Lモデルや Arx(GCG)10+7モデルなどから、特定のサブタイプの介在神経細胞が脆弱に減少する「インターニューロノパチー」という概念が確立されました。しかし、ヒト特有の皮質発達プロセスを完全には再現できないという種間差の限界も指摘されていました。
iPS細胞由来の脳オルガノイドが示した「予想外」の事実
この限界を打破したのが、患者由来のiPS細胞から作成したヒト脳オルガノイド(大脳皮質オルガノイドと神経節隆起オルガノイドを人為的に融合させたアセンブロイドモデル)を用いた研究です。ポリアラニン伸長(PAE)変異を持つオルガノイドで観察されたのは、それまでのマウスモデルから予想されていたのとは真逆の現象でした。
⚡ パラダイムシフト:マウスでは「停止」、ヒトでは「加速」
マウスモデルでは、ARXの機能不全により介在神経細胞の遊走は「停止・遅延」すると考えられてきました。しかしヒトのPAEオルガノイドでは、変異した介在神経細胞は細胞自律的に「加速して遊走する」ことが判明したのです。原因は、変異ARXがCXCR4プロモーターへの結合を異常に高めて受容体を過剰発現させること。結果として大脳皮質側のリガンドCXCL12への感受性が病的に高まり、神経細胞が異常な高速度で引き寄せられてしまいます。これらの細胞は最終的に皮質ネットワークに正しく組み込まれず、過同期的な神経活動(てんかん発作の細胞レベルでの実体)が生まれます。
🔬 ARX変異による介在神経細胞の遊走異常と治療標的
CXCR4-CXCL12軸の異常活性化が病態の中核。AMD3100で正常化が可能
ARX → CXCR4を適切に抑制
介在神経細胞は正常な速度で大脳皮質へ遊走
変異ARX → CXCR4が過剰発現
CXCL12への感受性が暴走、遊走が病的に加速
CXCR4を選択的にブロック
変異細胞の遊走速度が正常レベルに回復
健常細胞には影響を与えず、病的な過剰シグナルだけを選択的に遮断できる点が画期的
細胞レベルでの分子メカニズムをより詳細に視覚化したのが下の図です。受容体(CXCR4)の数の変化と、それに伴う化学走性応答の暴走、そしてAMD3100による選択的遮断の様子が表現されています。
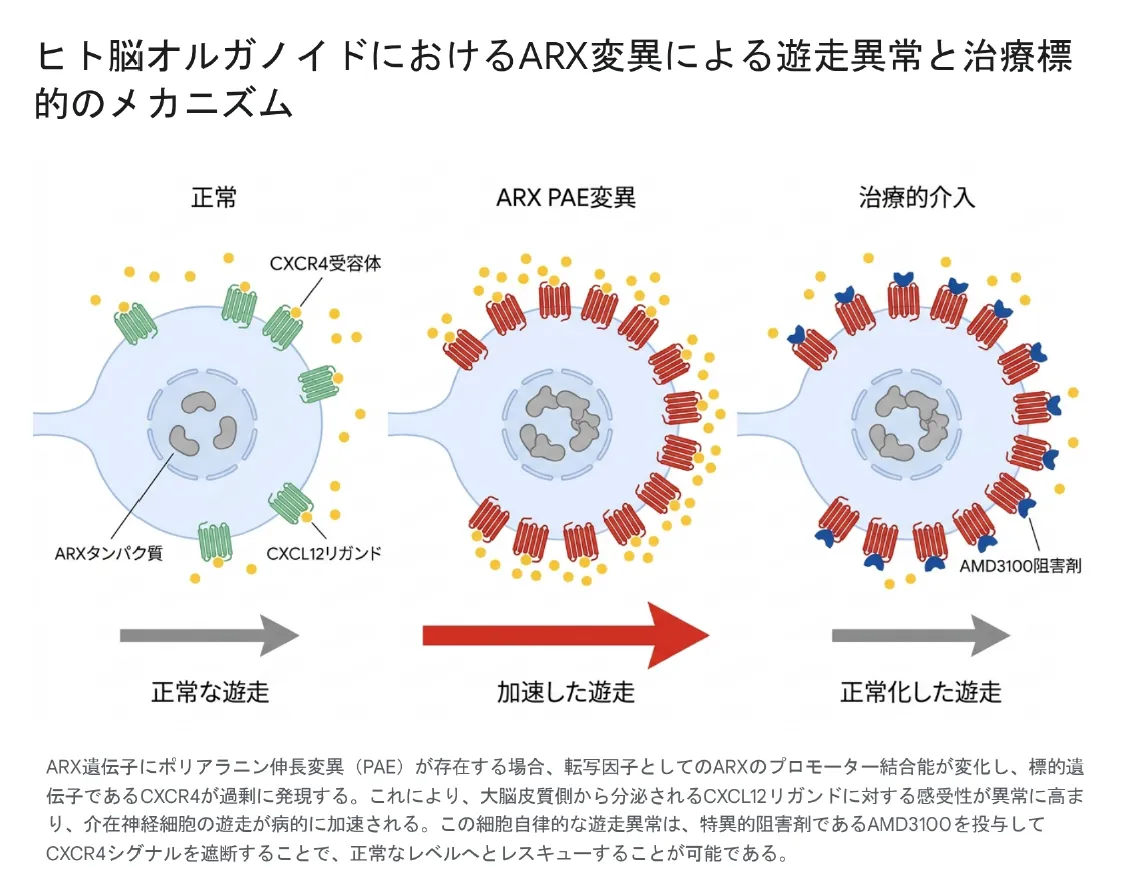
ARX遺伝子にポリアラニン伸長変異(PAE)が存在する場合、転写因子としてのARXのプロモーター結合能が変化し、標的遺伝子であるCXCR4が過剰に発現する。これにより、大脳皮質側から分泌されるCXCL12リガンドに対する感受性が異常に高まり、介在神経細胞の遊走が病的に加速される。この細胞自律的な遊走異常は、特異的阻害剤であるAMD3100を投与してCXCR4シグナルを遮断することで、正常なレベルへとレスキューすることが可能である。
この研究により、ARX-PAEアセンブロイドが発生のごく初期から過剰活動と同期性を示すことも判明し、病態の起源は胎生期にまで遡ることが実証されました。これは早期治療介入の重要性を裏付ける決定的な知見です。
8. 治療開発の最前線
これまでのARX関連てんかんに対する治療は、抗てんかん薬による発作の対症療法に依存してきました。しかし患者の多くは既存薬に抵抗性を示し、たとえ発作をコントロールできても根底にある知的障害や運動障害の進行は止められません。現在、病態の根本に介入する「疾患修飾治療(Disease-Modifying Therapies)」の開発が急速に進んでいます。
💊 AMD3100(Plerixafor)の薬剤再利用
既に造血幹細胞動員薬として承認済みのCXCR4選択的阻害剤。PAEオルガノイドで変異細胞の異常遊走を完全レスキュー。健常細胞には影響しない選択性の高さが期待されています。
🧬 アンチセンスオリゴヌクレオチド(ASO)
変異mRNAに相補的に結合する短い合成核酸。c.428_451dup24などの伸長変異に対して、変異アレルだけを選択的にノックダウンまたは異常エクソンをスキップさせる設計が可能です。MECP2重複症候群やHNRNPH2関連障害で前臨床成功例があります。
🦠 AAVベクター遺伝子治療
アデノ随伴ウイルスを介して正常ARX遺伝子を補充するアプローチ。SMAやDravet症候群で実用化が進んでいます。ただしARXは過剰発現も毒性を持ち得るため、内在性プロモーター制御の精密な再現が技術的課題です。
✂️ CRISPR次世代ゲノム編集
プライムエディティングやベースエディティングを用いれば、24bp重複配列や特定の点突然変異を二本鎖切断なしに直接修正できる可能性があります。Casgevyの承認以降、生体内ゲノム編集の臨床応用が加速しています。
細胞治療:抑制性介在神経細胞の直接移植
ARX関連疾患の本質が「GABA作動性介在神経細胞の喪失・機能不全による脱抑制」であるならば、外部から正常な抑制性ニューロンを直接補うアプローチは極めて論理的です。Neurona Therapeutics社が開発した「NRTX-1001」は、ヒト幹細胞から分化させた抑制性介在神経細胞を脳の発作焦点に直接移植する細胞治療薬で、薬剤抵抗性の内側側頭葉てんかんを対象とした第3相臨床試験(EPIC試験)に進んでいます。組織を切除する従来手術と異なり、機能を温存・修復する画期的アプローチとして、ARX関連難治性てんかんへの将来的な適応拡大が注目されています。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 ARX遺伝子・神経発達障害のご相談
ARX関連疾患をはじめとする希少遺伝性疾患・X連鎖性遺伝疾患のキャリア相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] Friocourt G, Parnavelas JG. Mutations in ARX result in several defects involving GABAergic neurons. Front Cell Neurosci. 2010;4:4. [Frontiers]
- [2] Marsh ED, Golden JA. Developing Models of Aristaless-related homeobox mutations. Jasper’s Basic Mechanisms of the Epilepsies [Internet]. 2012. [NCBI Bookshelf]
- [3] Shoubridge C, Fullston T, Gécz J. ARX spectrum disorders: making inroads into the molecular pathology. Hum Mutat. 2010;31(8):889-900. [PubMed]
- [4] Stromme P, et al. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat Genet. 2002;30(4):441-445. [Hum Mol Genet]
- [5] Friocourt G, et al. ARX revisited: involved in the development of GABAergic interneurons. Front Cell Dev Biol. 2025;13:1563515. [Frontiers]
- [6] Poirier K, et al. Genotype-phenotype associations for ARX gene duplication in X-linked mental retardation. Neurology. 2006;67(8):1473-1475. [Neurology]
- [7] Kato M, et al. A Longer Polyalanine Expansion Mutation in the ARX Gene Causes Early Infantile Epileptic Encephalopathy with Suppression-Burst Pattern (Ohtahara Syndrome). Am J Hum Genet. 2007;81(2):361-366. [PMC1950814]
- [8] Wallerstein R, et al. Ohtahara syndrome in a family with an ARX protein truncation mutation (c.81C>G/p.Y27X). Eur J Hum Genet. 2010. [PMC2987188]
- [9] Dual effects of ARX poly-alanine mutations in human cortical and ganglionic eminence assembloids. bioRxiv. 2024. [bioRxiv]
- [10] Marsh ED, et al. Arx expansion mutation perturbs cortical development by augmenting apoptosis without activating innate immunity. Dis Model Mech. 2020;13(3):dmm042515. [Dis Model Mech]
- [11] Neurona Therapeutics. Phase 3 EPIC Study for NRTX-1001 Cell Therapy in Epilepsy. 2025. [Neurona]
- [12] OMIM #300382. ARX, Aristaless-Related Homeobox, X-Linked. Johns Hopkins University. [NCBI Gene]






