目次

📍 クイックナビゲーション
CREBBP遺伝子は、DNAの「読まれ方」を切り替えるエピジェネティクスの司令塔ともいえる遺伝子です。この遺伝子が生まれつき変化しているとルビンスタイン・テイビ症候群やメンケ・ヘネカム症候群といった先天性の病気が起こり、一方で、生まれた後に体の一部の細胞で変化すると悪性リンパ腫や白血病の引き金になります。同じ一つの遺伝子が、まったく性質の異なる二つの病気の世界に関わっている——それがCREBBPの最大の特徴です。
Q. CREBBP遺伝子とは、ひとことで言うとどんな遺伝子ですか?
A. DNAの巻き付きをゆるめて遺伝子のスイッチを「オン」にする酵素(ヒストンアセチル化酵素)の設計図であり、たくさんの転写因子を束ねる巨大な「足場」でもある重要な遺伝子です。生まれつきの変化はルビンスタイン・テイビ症候群やメンケ・ヘネカム症候群を、後天的な変化はリンパ腫・白血病を引き起こすことがあります。
- ➤基本情報 → 16番染色体16p13.3に位置(HGNC:2348/NCBI Gene:1387/OMIM遺伝子番号600140)、別名CBP・KAT3A
- ➤分子の働き → ヒストンアセチル化酵素(HAT)+転写の足場。p53やBCL6など多数のタンパク質も調節
- ➤先天性疾患 → ルビンスタイン・テイビ症候群、メンケ・ヘネカム症候群、16p13.3欠失/重複症候群
- ➤がんとの関わり → 濾胞性リンパ腫・びまん性大細胞型B細胞リンパ腫・白血病。免疫逃避にも関与
- ➤最新研究 → 「合成致死性」を突くp300/CBP阻害剤(イノブロディブ等)の臨床開発が進行中
1. CREBBP遺伝子とは:基本情報
CREBBP(読み方は「シーレブビーピー」)は、CREB binding protein(CREB結合タンパク質)という意味の名前を持つ遺伝子です。タンパク質としての通称はCBP、酵素としての分類名はKAT3Aとも呼ばれます。16番染色体の短い腕の先端付近(16p13.3)に位置し、全身のほとんどすべての細胞で働いている、生命活動に欠かせない遺伝子です。
承認シンボル CREBBP/別名 CBP・KAT3A・RSTS/遺伝子座 16p13.3/HGNC:2348/NCBI Gene:1387/Ensembl:ENSG00000005339/遺伝子OMIM番号 600140
CBPはもともと、細胞内の信号を伝えるメッセンジャー役のタンパク質(CREB)にくっつく核内タンパク質として見つかりました。その後の研究で、CBPは単なる「くっつき役」ではなく、遺伝子のスイッチを入れる作業の中心にいる司令塔であることがわかってきました。CBPには非常によく似た「きょうだい遺伝子(パラログ)」としてEP300(p300)があり、構造も働きもよく似ています。この二つが互いを補い合う関係にあることが、後で述べる最新のがん治療のカギになります。
💡 用語解説:転写共役因子(てんしゃきょうやくいんし)
遺伝子が「読まれて」タンパク質がつくられる過程を転写といいます。転写を始めるには、DNAの特定の場所に結合する「転写因子」が必要ですが、転写因子だけでは力不足で、それを助ける補佐役が要ります。この補佐役が転写共役因子(コアクチベーター)です。CBPは数百種類もの転写因子を助ける万能の補佐役で、いわば「現場をまとめる総監督」のような存在です。
2. 遺伝子の働き:エピジェネティクスの司令塔
CBPの働きは大きく二つに分けられます。ひとつは「化学反応を起こす酵素としての働き」、もうひとつは「たくさんのタンパク質を集める足場としての働き」です。
💡 用語解説:エピジェネティクスとクロマチン
DNAの文字(塩基配列)そのものを変えずに、遺伝子の「読まれ方」を切り替える仕組みをエピジェネティクスといいます。細胞の中でDNAはヒストンという糸巻きにきつく巻き付いて(この状態をクロマチンといいます)コンパクトに収納されています。きつく巻かれた部分の遺伝子は「オフ」、ゆるんだ部分は「オン」になります。CBPはこの巻きのゆるめ役なのです。
働き① ヒストンアセチル化酵素(HAT)として遺伝子のスイッチを入れる
CBPの最も重要な働きが、ヒストンアセチル化酵素(HAT:Histone Acetyltransferase)としての活性です。CBPはヒストンの特定の部分(H3K18、H3K27、H3K56などのリジンというアミノ酸)に「アセチル基」という小さな目印を付けます。すると、ヒストンが帯びていたプラスの電気が打ち消され、マイナスの電気を持つDNAとの引き合う力が弱まります。その結果、きつく巻かれていたDNAがゆるんで、遺伝子が読まれやすい状態(オープンクロマチン)に変わります。CBPは、細胞の運命を決める重要な遺伝子のスイッチを「オン」にする最初の一手を担っているのです。
働き② ヒストン以外のタンパク質も調節する「分子スイッチ」
CBPはヒストンだけでなく、多くの重要なタンパク質にもアセチル基を付け、その働きを切り替える「分子スイッチ」として機能します。代表的な相手が、「ゲノムの守護神」と呼ばれるがん抑制タンパク質p53と、B細胞のがん遺伝子として知られるBCL6です。CBPがp53にアセチル基を付けると、p53はDNA損傷を見つけて細胞にブレーキをかける力を発揮します。一方、BCL6にアセチル基を付けると、その働きは逆に抑えられます。この「アクセル(p53)を入れ、ブレーキ役の暴走(BCL6)を抑える」という調節が、後で述べるがんの発生と深く関わってきます。
働き③ 巨大な「足場」として多くの経路をまとめる
CBPは酵素であると同時に、TAZ1・KIX・ブロモドメイン・TAZ2・ZZ・ID4など多くの結合用パーツを持つ巨大な足場タンパク質でもあります。これらのパーツを使って、CBPはさまざまな転写因子や基本的な転写の装置を一カ所に集め、遺伝子を読み始める準備を整えます。発生・成長・代謝・免疫・記憶の形成など、生命のほぼすべての主要な経路にCBPが関与しているのはこのためです。実際、CBPやEP300を完全に欠いたマウスは胎児の段階で生きられないことがわかっており、このタンパク質が生命の土台に不可欠であることを物語っています。
3. CREBBPの異常が引き起こす「2つの病気の世界」
CREBBPの異常は、いつ・どこで起こるかによって、まったく性質の異なる二つのタイプの病気につながります。これを理解することが、この遺伝子を知るうえでとても大切です。
① 生まれつきの変化 → 先天性疾患
受精卵の時点から体のすべての細胞にある変化(生殖細胞系列の変異)です。発生のプログラムが広く影響を受け、ルビンスタイン・テイビ症候群やメンケ・ヘネカム症候群などの先天性疾患が起こります。
② 生まれた後の変化 → がん
生まれた後に、体の一部の細胞だけで後天的に起こる変化(体細胞変異)です。これは遺伝するものではなく、悪性リンパ腫や白血病といった血液のがんの強力な引き金になります。
💡 用語解説:生殖細胞系列の変異と体細胞変異
生殖細胞系列の変異は、精子・卵子の段階や受精直後に生じ、体のすべての細胞に受け継がれます。次の世代に遺伝する可能性があります。
体細胞変異は、生まれた後に体の一部の細胞で偶然起こる変化で、その細胞とそこから分かれた細胞だけが持ちます。基本的に子どもには遺伝しません。同じCREBBPの変化でも、このどちらで起こるかによって、結果として現れる病気がまったく違ってくるのです。
4. CREBBPに関連する先天性疾患
かつてCREBBPの変異といえば「ルビンスタイン・テイビ症候群」を指すと考えられていました。しかし次世代シーケンサーの普及により、遺伝子の「どこ」に「どんな種類」の変化が起こるかによって、まったく違う病気になることがわかってきました。これは臨床遺伝学における大きな発見です。
ルビンスタイン・テイビ症候群(RTS)
ルビンスタイン・テイビ症候群(RTS1、OMIM #180849)は、CREBBP遺伝子全体の働きが失われる機能喪失型変異や、遺伝子を含む領域の欠失によって起こる常染色体顕性(優性)遺伝の先天性疾患です。CREBBPの変異が原因の約55〜60%を占め、きょうだい遺伝子EP300の変異によるものはRTS2(OMIM #613684)と分類されます。
💡 用語解説:ハプロ不全(ハプロふぜん)
私たちは遺伝子を父と母から1本ずつ、計2本持っています。ハプロ不全とは、片方の遺伝子が壊れて働かなくなり、残った1本だけでは必要な量のタンパク質をまかなえなくなる状態のことです。RTSでは、CREBBPの片方が壊れることでCBPタンパク質が正常の半分しかつくられず、これが胎児期の広い範囲の発生プログラムに影響します。
RTSの最も特徴的な身体的サインは「幅広の親指と足の親指(母趾)」です。そのほか、低身長、小頭症、中等度から重度の知的障害、しかめ笑いのような特徴的な顔つきなどがみられます。なお、16p13.3という領域が広く欠失すると、CREBBPだけでなく近くの複数の遺伝子もまとめて失われ、極端な成長障害や重い臓器の異常、繰り返す重症感染症を伴う重症型(16p13.3欠失症候群)となることがあります。
メンケ・ヘネカム症候群(MKHK)
メンケ・ヘネカム症候群(MKHK1、OMIM #618332)は、2010年代後半に初めて報告された非常にまれな常染色体顕性(優性)疾患です。RTSが「遺伝子全体の量が半分になる」病気であるのに対し、MKHKはCREBBPのエクソン30または31という特定の場所に起こるミスセンス変異によって生じます。
💡 用語解説:ミスセンス変異と新生突然変異(de novo)
ミスセンス変異は、DNAの文字が1つ変わって、設計図の一部のアミノ酸が別の種類に置き換わる変化です。タンパク質の形や働きが変わります。
新生突然変異(de novo)は、両親には存在せず、子どもに初めて生じた変化のこと。これらの先天性疾患の多くは新生突然変異で起こるため、ご両親が健康でも発症することがあります。
MKHKでは、タンパク質全体が壊れるのではなく、特定の相手(p53など)とのやりとりだけが選択的に変わったり、変異タンパク質が新しい性質(機能獲得や正常タンパク質の邪魔をする優性阻害効果)を持ったりすると考えられています。臨床的に最も重要な違いは、RTSの決め手だった「幅広の親指」がMKHKには見られないことです。広い額、両目の間隔が広い(眼距開離)、非常に長いまつげ、尖った顎など、RTSとは異なる独特の顔つきを示します。
4つの先天性疾患の比較
| 疾患 | 遺伝子異常の性質 | 指・趾の所見 | 主な特徴 |
|---|---|---|---|
| RTS1 | 機能喪失・微小欠失(ハプロ不全) | 著しく幅広の親指・母趾 | 低身長、小頭症、中〜重度の知的障害、特徴的顔貌 |
| 重症型RTS(16p13.3欠失) | 複数遺伝子の広範な欠失 | 幅広の親指・母趾 | 極端な成長障害、重篤な臓器異常、反復性感染症 |
| MKHK1 | エクソン30・31のミスセンス変異 | 正常な親指(幅広にならない) | 発達遅滞、長いまつげ・尖った顎などの独特の顔貌 |
| 16p13.3重複症候群 | 遺伝子の重複(過剰なコピー) | 異常な形の親指、関節拘縮 | 知的障害、心臓・生殖器・眼の異常 |

5. がんとの関わり:リンパ腫と白血病
生まれた後に体の一部の細胞で起こる体細胞変異としてのCREBBP異常は、血液のがんの強力なドライバー(推進役)になります。特に、リンパ節の中で抗体をつくる訓練をする場所「胚中心(はいちゅうしん)」から生まれる濾胞性リンパ腫(FL)やびまん性大細胞型B細胞リンパ腫(DLBCL)では、CREBBPに非常に高い頻度で変異が見つかります。
リンパ腫におけるCREBBPの異常は、主にHAT(アセチル化酵素)の働きを弱める機能喪失型の変化です。これによって細胞では次の3つのことが同時に起こり、がん化が一気に進みます。
- ➤分化のストップ:細胞を成熟させる遺伝子のスイッチが入らなくなり、未熟なまま激しく増え続けます。
- ➤ブレーキ役の暴走と守護神の麻痺:増殖を促すBCL6が抑えられなくなり、がんを抑えるp53も働けなくなります。
- ➤免疫からの隠れ身:免疫の目印(MHC-II)が細胞表面から消え、T細胞に見つからなくなります(免疫逃避)。
💡 用語解説:免疫逃避(めんえきとうひ)とMHC-II
私たちの体では、免疫のT細胞ががん細胞の表面にある「目印(MHC-IIという分子に乗せた抗原)」を見て、敵かどうかを判断します。CREBBPの変異でこの目印をつくる司令塔(CIITA)が止まると、目印が表面から消え、がん細胞が免疫の監視から「隠れる」ことができてしまいます。これを免疫逃避といい、リンパ腫の発生のかなり初期に起こる重要な出来事であることがわかっています。
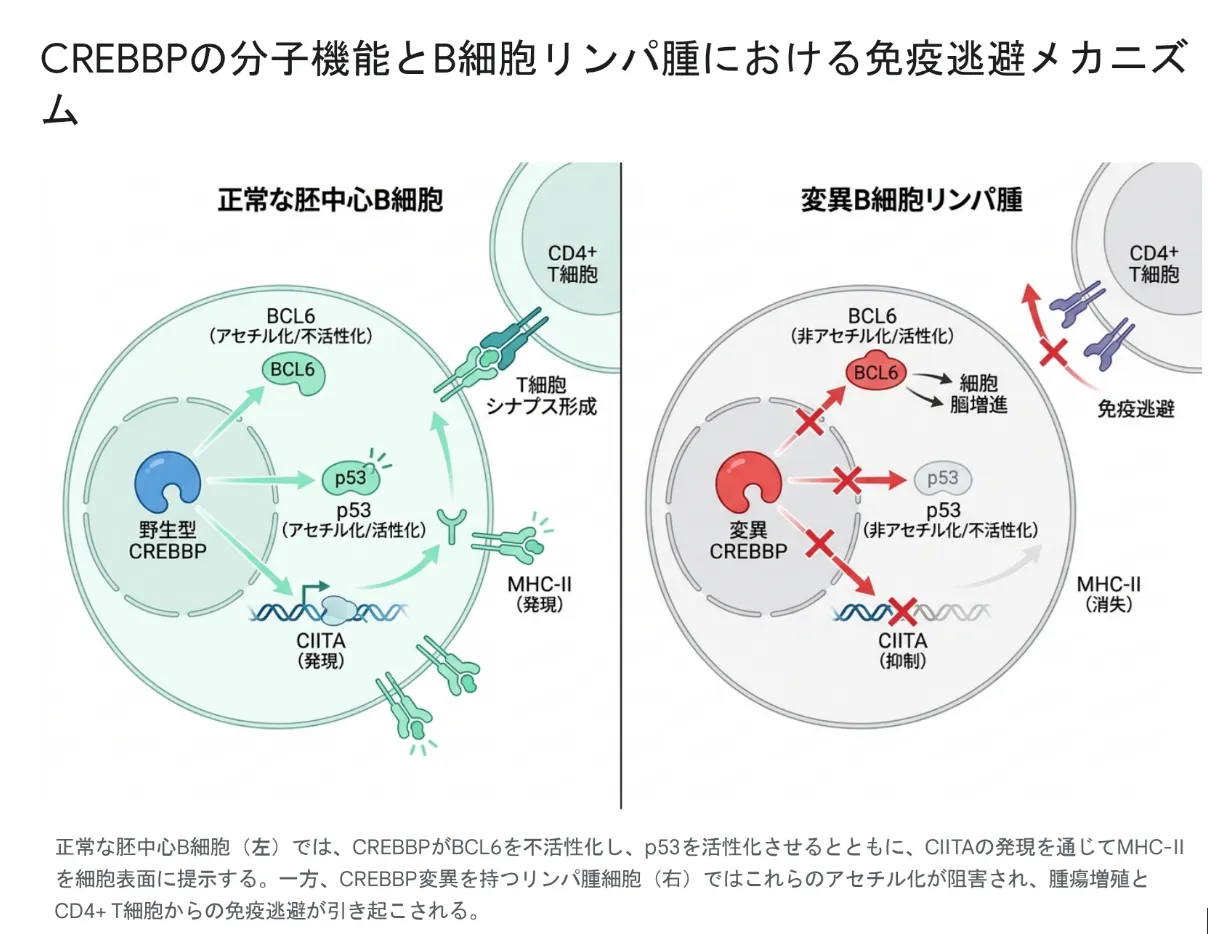
正常な胚中心B細胞(左)では、CREBBPがBCL6を抑え、p53を活性化し、CIITAを通じてMHC-IIを細胞表面に提示します。一方、CREBBP変異を持つリンパ腫細胞(右)ではこれらのアセチル化が妨げられ、腫瘍の増殖とT細胞からの免疫逃避が引き起こされます。
白血病における役割:転座と薬剤抵抗性
骨髄で起こる白血病でもCREBBPは病態の中心に関わります。急性骨髄性白血病(AML)の一部では、8番染色体と16番染色体が入れ替わる転座 t(8;16) によって、KAT6AとCREBBPがつながった異常なタンパク質(KAT6A-CREBBP融合)ができ、正常な血液細胞への成熟が止まってしまいます。このタイプはまれですが、予後が厳しいことで知られています。また、ほかのがんの化学療法のあとに二次的に生じる「治療関連白血病」では t(11;16) という転座も報告されています。
小児の急性リンパ性白血病(ALL)では、CREBBPの変異が「再発」の最も多い原因のひとつとして注目されています。ALLの治療の柱であるステロイド薬(グルココルチコイド)は、CBPを呼び寄せてがん細胞に自滅のスイッチを入れることで効きます。ところがCREBBPに変異があると、このスイッチが入らず、ステロイドが効かなくなって(抵抗性)再発の引き金になるのです。再発したALL患者の約18〜21%にこの変異が見つかる一方、寛解を保てた患者ではほとんど見られないことが報告されています。興味深いことに、これらの抵抗性細胞は別のエピジェネティック薬(HDAC阻害剤)には強く反応することがわかってきており、新たな治療の手がかりになっています。
6. 最新の治療研究:合成致死性とp300/CBP阻害剤
CREBBPの変異はがんを悪化させますが、逆説的に、がん細胞だけが持つ「弱点」も生み出します。この弱点を突くのが「合成致死性(synthetic lethality)」という考え方で、いま最も注目される標的治療の戦略です。
💡 用語解説:合成致死性(ごうせいちしせい)
2つの遺伝子のうち、どちらか片方が壊れても細胞は生きられるのに、両方そろって壊れると細胞が死ぬ——この現象を合成致死性といいます。CBP(CREBBP)とそのきょうだいEP300は働きがよく似ていて、互いを補えます。CREBBPが変異したがん細胞は、残ったEP300に頼って生き延びています。そこでEP300を薬で止めると、補い手を失ったがん細胞だけが死に、正常細胞は生き残る——という狙い撃ちが可能になります。
CREBBPとEP300の合成致死性アプローチ
(CREBBP正常)
✓ EP300(活性)
→ 生存
✗ EP300(阻害)
→ 生存(CREBBPが補う)
(CREBBP変異)
✓ EP300(活性)
→ 生存(EP300に依存)
✗ EP300(阻害)
→ 死滅/合成致死
正常細胞はCREBBPとEP300の両方が働くため、EP300を止められても生き残ります。一方、CREBBPが変異したがん細胞はEP300に頼っているため、EP300阻害剤を投与するとアセチル化の力が尽き、がん細胞だけが死ぬ(合成致死)よう設計されています。
開発が進む新薬:イノブロディブ(CCS1477)など
この原理を臨床に応用した代表が、イギリスのCellCentric社が開発するイノブロディブ(Inobrodib、開発コードCCS1477)です。飲み薬として使えるp300/CBP阻害剤で、これまでに固形がん・血液がんの患者500名以上で安全性と有効性が評価されてきました。特に、再発・難治性の多発性骨髄腫を対象とした第2相試験「DOMMINO-1(NCT07096778)」が進行中で、2025年12月の米国血液学会(ASH)では良好な用量最適化データが報告されています。2026年5月には2億2,000万ドル規模の大型資金調達が完了し、第3相試験への移行が予定されるなど、実用化に向けた期待が高まっています。
このほか、基礎研究で広く使われる選択的p300/CBP阻害剤A-485や、EP300だけを細胞内から分解して取り除く次世代技術PROTAC(タンパク質分解誘導薬)の開発も進んでいます。患者一人ひとりの遺伝子変化に合わせて最適な薬を選ぶ「精密エピジェネティック医療」が、血液がんの新しい標準治療として確立されていくことが期待されています。
7. CREBBPの遺伝子検査でわかること
CREBBPに関連する病気を調べる検査は、「生まれる前(出生前)」と「生まれた後(出生後)」で分けて考える必要があります。「診断=出生前」という誤解を避けることが大切です。
出生後の検査(生まれた後にお子さんを調べる)
症状からCREBBP関連疾患が疑われる場合、原因遺伝子を調べる遺伝子パネル検査が用いられます。ミネルバクリニックでは、CREBBPを含む以下の検査メニューをご用意しています。
- ➤ルビンスタイン・テイビ症候群NGSパネル検査:CREBBP・EP300・SRCAPの3遺伝子を一度に調べます。
- ➤低身長遺伝子パネル検査:CREBBPを含む低身長に関わる多数の遺伝子を調べます。
- ➤自閉症遺伝子検査:CREBBPを含む発達に関わる多数の遺伝子を網羅的に調べます。
出生前の検査(赤ちゃんが生まれる前に調べる)
CREBBP関連疾患の多くは新生突然変異(de novo)で起こるため、ご両親に変異がなくても赤ちゃんに生じることがあります。こうした単一遺伝子の新生突然変異を出生前に調べる選択肢として、母体の採血で行うNIPTインペリアルプランがあり、CREBBPもその対象遺伝子に含まれています。なお、出生前の確定診断には羊水検査・絨毛検査が必要です。
8. 遺伝カウンセリングの役割
CREBBP関連疾患の診断がついた後、あるいは検査を検討する段階で、遺伝カウンセリングが大きな支えになります。主に次のような内容を扱います。
- ➤遺伝形式と再発リスク:多くは新生突然変異で両親に変異はありませんが、常染色体顕性(優性)遺伝のため、患者本人が子どもを持つ場合の遺伝確率は理論上50%です。生殖細胞モザイクの可能性もあり、次のお子さんの検査についても相談できます。
- ➤見通しと支援の組み立て:RTSとMKHKでは知的障害の程度や合併症が異なります。正確な診断は、教育・医療・生活支援を計画するうえで欠かせません。
- ➤検査結果の解釈:CREBBPに変異が見つかっても、その「場所と種類」によって診断名が変わります。臨床遺伝専門医による精密な読み解きが重要です。
9. よくある誤解
誤解①「CREBBP変異=ルビンスタイン・テイビ症候群」
変異の場所と種類が異なれば、メンケ・ヘネカム症候群など別の疾患になります。一律に同じ診断とは言えません。
誤解②「がんになる遺伝子だから遺伝する」
リンパ腫・白血病で起こるCREBBP変異は後天的な体細胞変異で、基本的に子どもには遺伝しません。先天性疾患の変異とは別物です。
誤解③「親が健康なら遺伝子の病気ではない」
CREBBP関連疾患の多くは新生突然変異(de novo)です。ご両親に変異がなくても、お子さんに新しく生じることがあります。
誤解④「変異の量が同じなら同じ病気」
RTSは「量が半分(ハプロ不全)」、がんやMKHKは「働きの質的な変化」が関わります。同じ遺伝子でもメカニズムは多彩です。
10. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 遺伝子検査・遺伝カウンセリングについて
CREBBP関連疾患をはじめとする遺伝性疾患のご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
参考文献
- [1] MedlinePlus Genetics. CREBBP gene. [MedlinePlus]
- [2] NCBI Gene. CREBBP CREB binding protein (Gene ID: 1387). [NCBI Gene]
- [3] OMIM #180849. Rubinstein-Taybi Syndrome 1 (RSTS1). Johns Hopkins University. [OMIM]
- [4] OMIM #618332. Menke-Hennekam Syndrome 1 (MKHK1). Johns Hopkins University. [OMIM]
- [5] Identification of a novel, pathogenic CREBBP variant in a patient with Menke-Hennekam syndrome. Front Genet. 2025. [Frontiers]
- [6] Zhang J, et al. The CREBBP Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B-cell Lymphoma. Cancer Discov. 2017. [PMC5386396]
- [7] KMT2D acetylation by CREBBP at enhancers in normal and malignant germinal center B cells. PNAS. 2023. [PNAS]
- [8] Genome-wide CRISPR screens reveal synthetic lethal interaction between CREBBP and EP300 in DLBCL. PMC. 2021. [PMC8080727]
- [9] CREBBP is a Major Prognostic Biomarker for Relapse in Childhood B-cell ALL. PMC. 2024. [PMC11892942]
- [10] OncoKB. Somatic KAT6A-CREBBP Fusion. Memorial Sloan Kettering. [OncoKB]
- [11] CellCentric. Clinical Trials (Inobrodib / CCS1477). [CellCentric]






