目次

ルビンシュタイン・テイビ症候群1型は、CREBBPという遺伝子の変化によって生まれつき起こる、とてもまれな病気です。幅広い親指・足の親ゆび、特徴的な顔つき、低身長、発達の遅れなどを主な特徴とし、出生10〜12.5万人に1人ほどとされています。原因の多くは、ご両親にはない「新しく生じた変化(新生突然変異)」です。このページでは、一般の方にもわかるように、原因・症状・診断・治療・遺伝の考え方を臨床遺伝専門医がやさしく解説します。
Q. ルビンシュタイン・テイビ症候群1型とは、どんな病気ですか?まず結論だけ知りたいです
A. CREBBP遺伝子の変化によって起こる、生まれつきの多発奇形・発達症候群です。幅広い親指と足の親ゆび、特徴的な顔つき、出生後の成長の遅れ、中等度から重度の知的障害を主な特徴とします。原因の多くは、ご両親にはない新しく生じた遺伝子の変化(新生突然変異)で、きょうだいに同じ病気が起こる確率は通常とても低いと考えられています。
- ➤疾患の定義 → OMIM 180849、頻度は出生10〜12.5万人に1人、常染色体顕性(優性)遺伝
- ➤原因と仕組み → CREBBP遺伝子(16p13.3)の変化。全体の約55〜75%を占める「クロマチノパシー」
- ➤主な症状 → 幅広い親指・第一趾、特徴的顔貌、低身長、知的障害、心疾患などの全身合併症
- ➤鑑別診断 → メンケ・ヘネカム症候群や他のクロマチノパシーとの見分け方
- ➤診断・治療 → 遺伝子のシーケンス解析+MLPA、対症療法と最先端のエピジェネティック治療研究
1. ルビンシュタイン・テイビ症候群1型とは:定義と歴史
ルビンシュタイン・テイビ症候群(Rubinstein-Taybi syndrome:RSTS)は、特徴的な顔つき、著しく幅広い親指・足の親ゆび、出生後の成長の遅れ、中等度から重度の知的障害を中心的な特徴とする、まれな生まれつきの病気です。発生頻度は出生10万〜12.5万人に1人ほどと推定されています。
この病気は原因となる遺伝子によって大きく2つのタイプに分けられます。16番染色体(16p13.3)にあるCREBBP遺伝子の変化が原因のものを「ルビンシュタイン・テイビ症候群1型(RSTS1/OMIM 180849)」、22番染色体にあるEP300遺伝子の変化が原因のものを「2型(RSTS2/OMIM 613684)」とよびます。全体の約55〜75%をCREBBP遺伝子による1型が占めており、この病気の中心的なタイプといえます。
💡 用語解説:常染色体顕性(優性)遺伝
「常染色体」とは、性別を決めるX・Y染色体以外の染色体のことです。「顕性(けんせい)」は以前「優性」とよばれていた言葉で、2本ある染色体のうち、どちらか1本に変化があるだけで症状が現れる遺伝のしかたを指します。RSTS1は常染色体顕性(優性)遺伝の形をとりますが、実際にはご両親には変化がなく、お子さんで初めて生じた「新生突然変異(de novo)」によって発症する場合がほとんどです。
かつてこの病気は、その手足の特徴から「幅広親指・第一趾症候群(Broad thumb-hallux syndrome)」とよばれていました。独立した症候群として確立したのは、複数の医師による観察の積み重ねによります。文献上もっとも古い報告は1957年、ギリシャの医師らによるものでした。その後、米国の小児科医ジャック・ルビンシュタイン博士が特徴的な顔つきと指の所見を示す子どもたちに共通点を見いだし、1963年に小児放射線科医のフーシャン・テイビ博士が一連の症例を正式に報告したことで疾患概念が確立し、両者の名をとって命名されました。
2. 原因遺伝子CREBBPと分子メカニズム
RSTS1の原因は、16番染色体の16p13.3という場所にあるCREBBP遺伝子の変化です。この遺伝子は、細胞の中で「遺伝子のスイッチ」を入れる役割をもつ、CBPというタンパク質の設計図です。RSTS1がなぜ全身のさまざまな臓器に影響をおよぼすのか——その答えは、CBPのもつ特別な働きにあります。
💡 用語解説:CREBBP遺伝子とCBPタンパク質
CREBBP(CREB結合タンパク質)は、約2,442個のアミノ酸からなる大きなタンパク質「CBP」をつくる遺伝子です。CBPは、たくさんの遺伝子のはたらきを同時に調節する「指揮者」のような存在で、細胞の成長・分化、胎児の正常な発生、出生後の記憶の形成などに欠かせません。ちなみに2型の原因であるEP300は、CBPとよく似た「p300(E1A結合タンパク質p300)」をつくる遺伝子で、両者は親戚のような関係にあります。
💡 用語解説:エピジェネティクスとクロマチノパシー
「エピジェネティクス」とは、DNAの文字(塩基配列)そのものは変えずに、遺伝子の「読まれ方(オン・オフ)」を調節する仕組みのことです。この調節の仕組みがこわれて起こる病気のグループを「クロマチノパシー」とよび、RSTS1はその代表的なモデル疾患とされています。RSTSはDNAの文字が壊れる病気ではなく、遺伝子の「読まれ方」の調節が乱れる病気なのです。
CBPの中心的なはたらきは、「ヒストンアセチル基転移酵素(HAT)」としての機能です。少し専門的になりますが、ここがRSTS1の病態のいちばん大事なポイントなので、図でやさしく見ていきましょう。
💡 用語解説:ヒストンアセチル化とHAT
DNAは「ヒストン」というタンパク質に巻きついてコンパクトに収納されています。CBP(HAT)はヒストンに「アセチル基」という小さな目印を付ける酵素で、これによってDNAの巻きつきがゆるみ、遺伝子が読み取られやすい状態(オープンクロマチン)になります。アセチル化=遺伝子をオンにする鍵とイメージするとわかりやすいです。
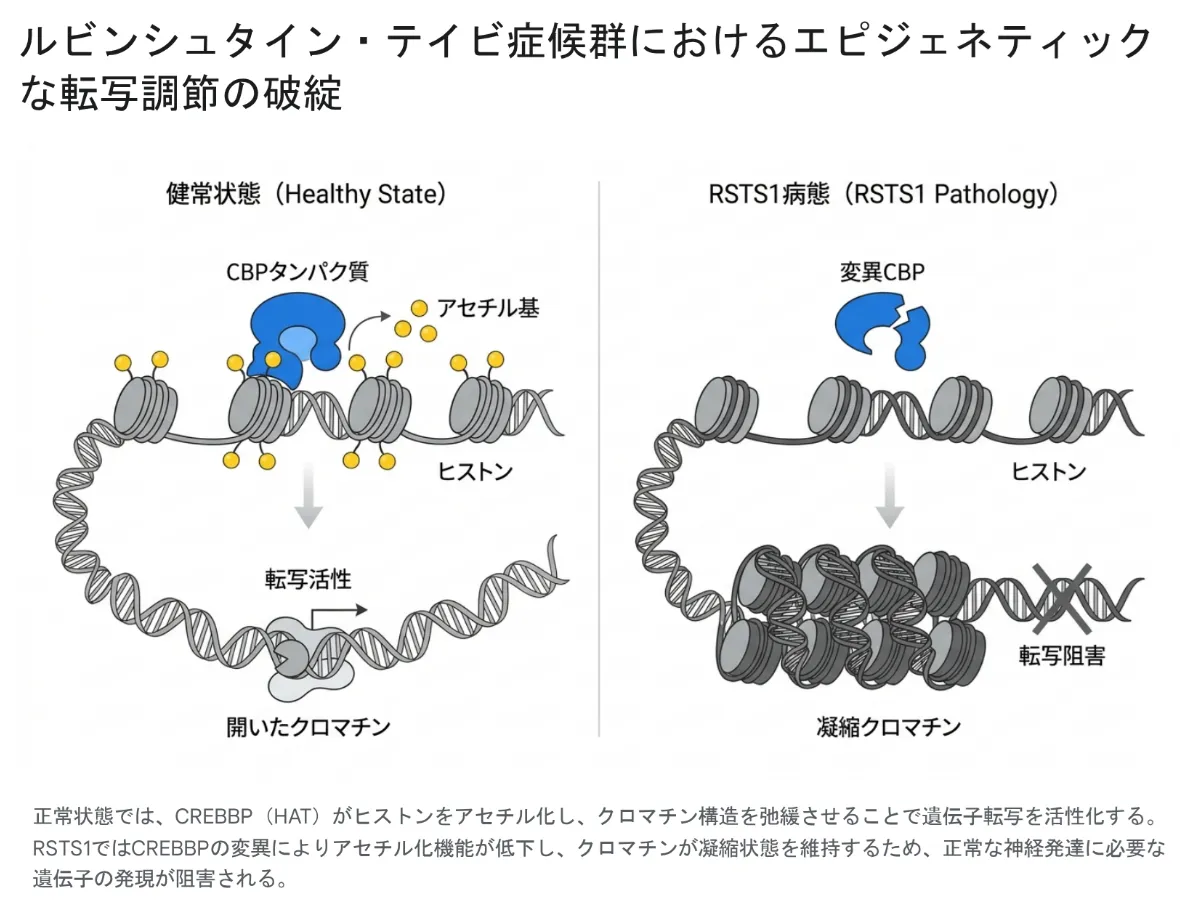
健常な状態ではCBP(HAT)がヒストンをアセチル化し、クロマチンを緩めて遺伝子の転写を活性化します。RSTS1ではCREBBPの変化によってアセチル化のはたらきが低下し、クロマチンが凝縮したまま維持されるため、正常な神経発達に必要な遺伝子の発現が妨げられます。
💡 用語解説:ハプロ不全(はぷろふぜん)
遺伝子は通常2本ペアで持っていますが、そのうち1本が働かなくなり、残り1本だけでは必要な量のタンパク質をまかなえない状態を「ハプロ不全」といいます。RSTS1では、片方のCREBBPの変化によってCBPが正常の約半分しか作られなくなることが、病気の中心的な原因と考えられています。
どんな遺伝子の変化が起こるのか
CREBBP遺伝子の変化のしかたはさまざまです。患者さんの約30〜50%は遺伝子の中の小さな変化(点変異)で、ミスセンス変異・ナンセンス変異・フレームシフト変異・スプライス部位変異などが含まれます。一方、約10〜20%は遺伝子の一部から全体に及ぶ大きな欠失(なくなること)です。さらにまれに、欠失がCREBBPだけでなく周囲の遺伝子まで巻き込む「16p13.3欠失症候群」とよばれる状態になり、古典的なRSTSよりも重い合併症を伴うことがあります。
💡 用語解説:ミスセンス変異・ナンセンス変異・新生突然変異
ミスセンス変異は、DNAの文字が1つ変わってアミノ酸が別の種類に置き換わる変化です。ナンセンス変異は、タンパク質づくりが途中で止まってしまう変化で、CBPの機能が失われる原因になります。
新生突然変異(de novo)とは、ご両親の精子・卵子、または受精直後に新たに生じた変化のことで、ご両親には同じ変化が見られません。RSTS1の約99%にはご家族の病歴がなく、この新生突然変異によって発症すると考えられています。
3. 主な症状と全身合併症
RSTS1の症状はとても多彩で、重さには大きな個人差があります。ここでは大きく4つの領域に分けて、特徴的な所見をご紹介します。
📏 成長
- 胎児期の成長は正常範囲内のことが多い
- 生後数か月で身長・体重・頭囲が急に伸び悩む
- 小頭症、成人期の著しい低身長
- 小児期後半以降は肥満のリスク
👶 頭蓋顔面・口腔
- 外下方に下がる眼瞼裂、アーチ状の眉
- 凸状の鼻すじ、下方に垂れる鼻柱
- 笑うと目を細める独特の表情(しかめ笑い)
- 高口蓋、永久歯のタロン結節
🖐️ 骨格・四肢
- 幅広い親指・足の親ゆび(ほぼ全例)
- 指先がへら状に広がる
- 親指が内側に強く曲がる
- 第5指の斜指症、合指症を伴うことも
🧠 神経・発達
- 運動・言語のマイルストーンの遅れ
- 平均IQは35〜50(中等度〜重度の知的障害)
- 自閉症的行動・てんかんを伴うことも
- 記憶力や巧緻運動が比較的保たれる例も
💡 用語解説:タロン結節(たろんけっせつ)
永久歯(とくに上の前歯)の裏側にできる、ワシの爪に似た副次的な突起のことです。RSTS患者さんに非常に多くみられる歯の特徴で、すき間に汚れがたまりやすく、むし歯のリスクを高める要因にもなります。歯科での定期的なケアが大切です。
全身に及ぶ合併症
RSTS1では、複数の臓器にまたがる合併症がしばしばみられます。代表的なものを表にまとめました(横にスクロールできます)。
| 臓器系 | 主な所見 | 臨床的なポイント |
|---|---|---|
| 心血管 | 先天性心疾患(約1/3)。心房中隔欠損・心室中隔欠損・動脈管開存・大動脈縮窄など | 確定診断時に心エコー評価を行う |
| 泌尿生殖器 | 男児の停留精巣(ほぼ全例)、水腎症、尿路の重複・逆流 | 早期の外科的対応が必要なことがある |
| 呼吸器・睡眠 | 閉塞性睡眠時無呼吸、喉頭・気管軟化症、反復する気道感染 | 全身麻酔時に特別な注意が必要 |
| 眼・耳鼻 | 斜視、屈折異常、眼瞼下垂、難聴、反復性中耳炎、鼻涙管閉塞 | 聴覚スクリーニングを早期に |
| 消化器・皮膚 | 重度の便秘、哺乳障害、胃食道逆流、ケロイド形成 | 手術痕がケロイドになりやすい |
| 腫瘍リスク | 髄膜腫、毛母腫、横紋筋肉腫などが一般集団より生じやすい | 気になる症状は早めに相談を |
なお、狭い口蓋や筋緊張の低さから全身麻酔のリスクが高くなるため、大きな手術の前には睡眠の評価(ポリソムノグラフィ)を行い、麻酔科医と十分に連携することがとても大切です。これは手術を受けるすべてのRSTS患者さんに共通する重要な注意点です。

4. 鑑別診断:似ている病気との見分け方
RSTS1の幅広い症状は、ほかのいくつかの病気と似ていることがあります。とくに、遺伝子の「読まれ方」の調節に関わるクロマチノパシーの仲間や、染色体の病気との見分けが重要です。
メンケ・ヘネカム症候群との鑑別
同じCREBBP遺伝子が原因ですが、変化の起こる場所(エクソン30〜31)が異なり、発達遅滞や特有の顔つきを示す別の病気です。手足の特徴はRSTSほど目立たないことが多く、遺伝子検査で変化の部位を確認することが鑑別の鍵になります。
他のクロマチノパシーとの鑑別
ウィーデマン・シュタイナー症候群やボーリング・オピッツ症候群など、エピジェネティックな調節に関わる他の病気とも症状が重なることがあります。最終的には遺伝子検査で原因を特定します。
染色体の病気との鑑別
重い発達の遅れを伴う場合、ダウン症候群・ウォルフ・ヒルシュホーン症候群・ウィリアムズ症候群などの染色体の病気との区別も大切です。微小欠失症候群のスクリーニングが手がかりになります。
原因不明の重い知的障害や多発奇形の検査として「染色体マイクロアレイ(CMA)」が最初に行われた結果、事前に疑っていなくてもCREBBP領域の欠失が偶然見つかり、後からRSTSと診断されるケースも少なくありません。
5. 診断と遺伝子検査の進め方
RSTSの診断は、ていねいな臨床的評価から始まり、遺伝子検査で原因を特定することで確定します。2024年には世界で初めての国際コンセンサスステートメント(診断と管理に関する合意文書)が発表され、診断の考え方が整理されました。
専門家グループは、分子的に確定した多くの患者さんに共通してみられる所見——下がった眼瞼裂、下方に垂れた鼻柱、しかめ笑い、タロン結節、幅広く曲がった親指と足の親ゆび、停留精巣など——を「強く示唆する所見」として位置づけています。
遺伝子検査の段階的アプローチ
臨床的にRSTSが疑われる場合、まずCREBBP遺伝子のシーケンス解析で点変異を探します。変化が見つからなければ、MLPA法などで遺伝子の大きな欠失や重複を調べます。それでも見つからない場合はEP300遺伝子を同様に調べます。近年は、これらを一度に調べる次世代シーケンサーによるマルチジーンパネル検査も広く使われています。
💡 用語解説:MLPAと染色体マイクロアレイ(CMA)
MLPAは、遺伝子の一部(エクソン単位)の欠失や重複を精度よく調べる方法です。通常の染色体検査(Gバンド法)では見つけにくい小さな欠失も検出できます。染色体マイクロアレイ(CMA)は、ゲノム全体のコピー数の増減を一度に調べる検査で、原因不明の発達遅滞や多発奇形の出生後の精査で広く使われます。
出生後の検査(生まれたあとの原因検索)
生まれたあとに発達の遅れや特徴的な所見からRSTSが疑われる場合、血液などを用いた遺伝子検査で原因を調べます。CREBBPは、ミネルバクリニックの以下のパネル検査の対象遺伝子に含まれています。
- ➤発達障害・学習障害・知的障害遺伝子パネル検査(689遺伝子・CREBBPを含む)
- ➤自閉症スペクトラム障害遺伝子パネル検査(122遺伝子・CREBBPを含む)
出生前の検査(妊娠中の選択肢)
出生前の確定診断は、羊水検査・絨毛検査で行います。ご家族にすでに見つかっている変化がある場合は、確実な診断が可能です。なお、一般的なNIPT(新型出生前診断)は染色体の数の異常が主な対象で、CREBBPの小さな点変異を直接調べる検査ではありません。妊娠中に超音波で胆嚢の腫大や脳の所見などからRSTSが疑われることもあり、その際は遺伝カウンセリングを通じて検査の選択肢を整理します。
CREBBPを含む多数の遺伝子を対象に、お子さんに新しく生じた変化を出生前に調べる選択肢として、NIPTインペリアルプランがあります。ただしこれはスクリーニング検査であり、確定診断ではありません。どの検査が適しているかは状況によって異なりますので、まずは遺伝カウンセリングでご相談ください。
6. 治療と長期的な管理
RSTS1には根本的な治療法はまだなく、それぞれの合併症に対する対症療法と、生涯にわたる多職種チームによるサポートが基本になります。2024年の国際コンセンサスでは、重症度の評価を医師だけで決めるのではなく、ご本人とご家族を中心に行うべきであることが強調されています。
整形外科的な管理
親指の形の手術は通常3〜4歳ごろに検討されますが、RSTSの手術経験が豊富な専門医が行うことが推奨されます。思春期以降は膝蓋骨(膝のお皿)の脱臼に注意が必要で、放置すると歩行に大きな影響が出ることがあります。側弯症は約20%にみられますが、多くは進行性ではありません。
呼吸・睡眠・麻酔
気道の狭さや筋緊張の低さから閉塞性睡眠時無呼吸(OSA)のリスクが高く、成人の約25%に影響します。手術前には睡眠の評価を行い、麻酔の安全性を慎重に確認することが欠かせません。
発達支援・生活
早期からの療育・教育支援が、お子さんの力を伸ばすうえで重要です。知的障害の程度には差があり、適切な支援のもとで社会生活を送る方も多くいらっしゃいます。RSTSの多くは成人まで生存します。
研究が進むエピジェネティック治療
これまで対症療法が中心でしたが、近年、病気の根本である「遺伝子の読まれ方の障害」を直接ねらう新しい治療研究が急速に進んでいます。鍵となるのがHDAC阻害剤という考え方です。
💡 用語解説:HDAC阻害剤とは
アセチル基を「付ける」のがCBP(HAT)なら、アセチル基を「外す」のがHDACという酵素です。RSTS1ではアセチル化が不足しているので、外す側のHDACのはたらきをおさえれば、細胞の中のアセチル化のバランスを取り戻せるのではないか、という発想です。これがHDAC阻害剤による治療研究の基本的な考え方です。
動物実験では、大人になってからHDAC阻害剤を投与しても、記憶や学習の障害が改善することが示され、「知的障害は必ずしも一生変わらないものではないかもしれない」という希望につながりました。さらに、腸内細菌がつくる「酪酸(らくさん)」という物質にもHDACをおさえる作用があり、RSTS患者さんの腸では酪酸をつくる菌が減っていることもわかってきています。将来的に、食事やプロバイオティクスを使った副作用の少ない方法への応用が期待されています。
ただし、抗てんかん薬バルプロ酸を用いてヒトで行われた臨床試験では、記憶の改善について統計的に明確な効果は確認されませんでした。動物の成果をそのまま人に当てはめることの難しさを示しており、今後はより選択的な薬や、投与する時期の工夫が課題となっています。現時点では確立した薬物治療はなく、研究段階にあることを正しく理解しておくことが大切です。
7. 遺伝カウンセリングの意義
RSTS1の診断後は、ご家族へのていねいな遺伝カウンセリングが大切です。臨床遺伝専門医は、特定の検査や選択を押しつけることなく、中立の立場で情報を提供し、最終的な決定はご家族にゆだねます。
- ➤再発リスクの説明:多くは新生突然変異のため、きょうだいで再び起こる確率は通常1%未満と低いとされます。ただし、ご両親の生殖細胞モザイク(精子や卵子の一部だけに変化がある状態)の可能性は完全には否定できないため、次のお子さんの出生前診断についても説明します。
- ➤遺伝形式の説明:常染色体顕性(優性)遺伝のため、患者さんご本人がお子さんをもつ場合の遺伝確率は理論上50%です。
- ➤合併症管理の見通し:心臓・腎臓・聴覚・睡眠などの合併症を早めに把握し、計画的にフォローすることが予後を支えます。
- ➤心理的サポート:診断は数値や検査結果だけの問題ではなく、ご家族の将来に関わる大切な意思決定です。気持ちの面でも継続的に伴走します。
8. よくある誤解
誤解①「幅広い親指がなければRSTSではない」
症状の出かたには大きな幅があり、親指の特徴が目立たない方もいます。顔つき・発達・全身の合併症を総合して判断し、必要なら遺伝子検査で確認します。
誤解②「知的障害があるから何もできない」
知的能力の保たれ方には個人差があり、適切な支援のもとで社会生活を送る方も多くいます。動物実験では知的機能の改善も示されつつあり、研究も進んでいます。
誤解③「NIPTで必ずわかる」
一般的なNIPTは染色体の数の異常が主な対象で、CREBBPの小さな点変異は直接の対象ではありません。出生前の確定診断は羊水検査・絨毛検査で行います。
誤解④「親が健康だから遺伝ではない」
RSTS1の多くは新生突然変異で、ご両親には同じ変化がないことがほとんどです。「両親が健康だから遺伝とは関係ない」という思い込みが、診断を遅らせることがあります。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 希少疾患の診断・遺伝カウンセリングについて
ルビンシュタイン・テイビ症候群をはじめとする希少遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] OMIM. Rubinstein-Taybi Syndrome 1(RSTS1); #180849. Johns Hopkins University. [OMIM 180849]
- [2] Stevens CA. Rubinstein-Taybi Syndrome. GeneReviews®. University of Washington. [GeneReviews NBK1526]
- [3] MedlinePlus Genetics. Rubinstein-Taybi syndrome. National Library of Medicine. [MedlinePlus]
- [4] Diagnosis and management in Rubinstein-Taybi syndrome: first international consensus statement. J Med Genet. 2024;61(6):503-519. [J Med Genet]
- [5] CREBBP gene; CREB binding protein. OMIM *600140. Johns Hopkins University. [OMIM 600140]
- [6] Fetal phenotype of Rubinstein-Taybi syndrome caused by CREBBP mutations. Prenat Diagn / PubMed. 2019. [PubMed 30633342]
- [7] Insights into the Role of the Microbiota and of Short-Chain Fatty Acids in Rubinstein-Taybi Syndrome. Int J Mol Sci. 2021;22(7):3621. [MDPI IJMS]
- [8] Orphanet. Rubinstein-Taybi syndrome. ORPHA:783. [Orphanet]






