目次

📍 クイックナビゲーション
PTPN11遺伝子は、細胞の増殖や分化のアクセルとブレーキを調整する「SHP2」というタンパク質の設計図です。この一つの遺伝子の変化が、生まれつきのヌーナン症候群やLEOPARD症候群といった先天性疾患の原因にも、生まれた後に起こる白血病や一部のがんの引き金にもなります。本記事では、SHP2タンパク質のはたらきから関連する病気、そして出生前診断(NIPT)との関わりまでを、臨床遺伝専門医がやさしく解説します。
Q. PTPN11遺伝子とは何ですか?まず結論だけ知りたいです
A. PTPN11は、細胞の成長シグナルを調整する「SHP2」という酵素をつくる遺伝子です。生まれつき持っている変化(生殖細胞系列変異)はヌーナン症候群(原因の約50%)やLEOPARD症候群などの先天性疾患を、生まれた後に細胞で起こる変化(体細胞変異)は若年性骨髄単球性白血病(JMML)などのがんを引き起こします。多くは家族歴のない新生突然変異(de novo変異)で生じ、NIPTで出生前に調べられる場合があります。
- ➤SHP2の正体 → 12番染色体にある遺伝子がつくる、シグナルの「促進役」のホスファターゼ(脱リン酸化酵素)
- ➤分子のパラドックス → ヌーナンは「機能獲得」、LEOPARDは「機能喪失」なのに症状が似る不思議
- ➤がんとの関係 → JMMLの約35%のドライバー。固形がんでも体細胞変異として関与
- ➤遺伝のしかた → 常染色体顕性(優性)遺伝。大半は新生突然変異で、父親の年齢と関連
- ➤調べ方 → 出生前はNIPT・確定検査、出生後は遺伝子パネル検査。専門医の遺伝カウンセリングが前提
1. PTPN11遺伝子とSHP2タンパク質:基本のキ
PTPN11遺伝子は、ヒトの12番染色体の長腕(12q24.13)に位置する遺伝子で、「SHP2(Src homology 2 domain-containing protein tyrosine phosphatase-2)」というタンパク質の設計図になっています[1]。なお同じタンパク質は文献によってPTP2C・SH-PTP2・SH-PTP3・BPTP3などの別名でも呼ばれますが、いずれも同じPTPN11/SHP2を指します。SHP2はからだのほとんどの細胞の細胞質に存在し、細胞が「増えなさい」「ある役割を持つ細胞に成熟しなさい」「動きなさい」「生き続けなさい」といった指令を受け取り、伝える際の中継点として働いています[1]。
💡 用語解説:プロテインチロシンホスファターゼ(PTP)
細胞のなかでは、タンパク質に小さな「リン酸」という目印を付けたり外したりすることで、スイッチのオン・オフが切り替えられています。リン酸を付ける酵素を「キナーゼ」、外す酵素を「ホスファターゼ」と呼びます。SHP2はチロシンというアミノ酸からリン酸を外す「プロテインチロシンホスファターゼ」の一種です。ふつうホスファターゼはシグナルを「止める」役ですが、SHP2は例外的にシグナルを「促進する」という珍しい性質を持っています。
特に胎児が育つ過程で、SHP2は心臓の形づくり、血液細胞のもとになる造血、骨や結合組織の発達に欠かせない役割を担っています[1]。それだけに、この一つの遺伝子に変化が起こると、心臓・骨・血液・神経といった複数の臓器に同時に影響が出るのです。PTPN11は、ヒトのチロシンホスファターゼのなかで最初に明確に「がん原遺伝子(がんを引き起こしうる遺伝子)」と位置づけられた、医学的にも重要な遺伝子です[7]。
2. SHP2の分子構造と「自己阻害」という巧妙なしくみ
PTPN11の変異がなぜこれほど多彩な病気を起こすのかを理解する鍵は、SHP2タンパク質の「かたち(立体構造)」にあります。SHP2は大きく4つの領域からできています[2]。N末端側に、リン酸化されたチロシンを認識して結合する2つの「SH2ドメイン」(N-SH2とC-SH2)があり、中央に実際にリン酸を外す「PTP触媒ドメイン」、そして末端にSHP2自身の働きを調整するチロシン(Tyr542・Tyr580)を持つ「C末端テール」が続きます[2]。触媒ドメインの中心には、脱リン酸化反応を担う活性中心のシステイン(Cys459)があります[8]。
SHP2の最大の特徴であり、病気の理解の核心となるのが「自己阻害」というしくみです[2]。刺激がない普段の状態では、SHP2は折りたたまれた「閉じた(Closed)」かたちをとり、N-SH2ドメインが触媒ポケットに物理的にはまり込んで、酵素活性をオフにしています[2]。外から成長因子の刺激が届き、足場となるタンパク質のリン酸化チロシンにSHP2が結合すると、N-SH2が外れて触媒部位が現れ、「開いた(Open)」かたちに切り替わって初めてフルに働きます。この開いた状態を安定させる鍵として、アルギニン111(R111)とグルタミン酸249(E249)のあいだに生じるイオン対が重要であることも、最近の構造解析で示されています[4]。
💡 用語解説:自己阻害(オートインヒビション)
酵素が自分自身の一部でフタをして、必要なときだけ働くように制御するしくみです。SHP2では、N-SH2ドメインが触媒部位に「自分でフタをする」ことで、刺激がないときは静かにしています。安全装置のようなものだと考えてください。多くの病的変異は、この安全装置を壊してしまい、スイッチが入りっぱなしになることで病気を起こします。
3. SHP2が制御するシグナル伝達ネットワーク
SHP2は一つの経路だけでなく、細胞のなかの多くのシグナルの流れに関わります。最もよく知られているのが「RAS/MAPK経路」の促進です[2]。標的のリン酸化チロシンに結合したSHP2は、GRB2やSOSといった分子と大きな複合体をつくり、RAS→RAF→MEK→ERKというリレーに点火します。ERK経路が完全に活性化するには、SHP2のホスファターゼ活性そのものが不可欠であることが実験で確かめられています[2]。
💡 用語解説:RAS/MAPK経路
細胞が「増えろ」「成熟しろ」「動け」という指令を受け取り、核へ伝えるための主要な情報伝達ルートです。受容体(RTK)→ アダプター(SOS1・SHP2など)→ RAS → RAF → MEK → ERK と、バケツリレーのように順番にスイッチが入っていきます。心臓・骨・皮膚・脳など多くの臓器の発達に関わるため、この経路の異常はさまざまな先天性疾患の原因になります。
SHP2は血液をつくる造血の現場でも中心的です。ヒトの造血前駆細胞を使った研究では、SHP2のホスファターゼ機能を失わせると血球コロニーの形成が減り、最適な造血にはSHP2の酵素活性が根本的に必要であることが示されました[6]。さらにリンパ球では、IL-2やIL-15のシグナル伝達や、IL-6受容体gp130を介したERK活性化に関与し、自己免疫を起こしうるSTAT3の過剰な活性化を抑えるバランス役も果たします[5]。
近年とりわけ注目されているのが、免疫チェックポイント分子PD-1の抑制シグナルへの関与です。T細胞が疲弊するときにPD-1がSHP2を呼び込み、T細胞受容体(TCR)の活性化シグナルを脱リン酸化して免疫にブレーキをかけます[5]。この性質が、後で述べるがん治療におけるSHP2阻害薬の重要性につながっています。
4. 生まれつきの変異とRAS病:ヌーナン症候群とLEOPARD症候群
🔍 関連記事:ヌーナン症候群1型(PTPN11)/LEOPARD症候群1型
精子や卵子の段階から全身の細胞が持っている変異(生殖細胞系列変異)は、発生の初期からRAS/MAPK経路の調整を乱し、「RAS病(RASopathies)」と総称される先天性疾患を起こします。代表がヌーナン症候群と、多発性黒子を伴うLEOPARD症候群(NSML)です。
ヌーナン症候群(機能獲得型変異)
ヌーナン症候群は、特徴的な顔つき、翼状頸、低身長、先天性心疾患(肺動脈弁狭窄や肥大型心筋症など)、胸郭異常、出血傾向、さまざまな程度の発達の遅れを示す常染色体顕性(優性)遺伝の病気です[11]。患者さんの約50%でPTPN11変異が見つかり、その大半は1つのアミノ酸が別のものに置き換わるミスセンス変異です[11]。これらは自己阻害の界面を壊して安全装置を不安定にし、SHP2を「オンのまま」にする機能獲得型変異です[3]。
💡 用語解説:機能獲得型・機能喪失型
機能獲得型(GOF)は、変異によってタンパク質が本来より過剰に・余計に働いてしまうタイプ。機能喪失型(LOF)は、働きが弱くなる・なくなるタイプです。ふつうは「獲得なら活発、喪失なら不活発」と逆の症状になりそうですが、PTPN11ではこの常識が通用しない不思議な現象(パラドックス)が起こります。
LEOPARD症候群と「分子のパラドックス」
LEOPARD症候群(多発性黒子を伴うヌーナン症候群/NSML)は、ヌーナン症候群とよく似た顔つき・体つきに加え、全身の多数の黒子や、しばしばより重い肥大型心筋症を伴います[10]。約85%がPTPN11変異によるもので、Y279CとT468Mという特定の置換だけで約65%を占めます[3]。ここで長年の謎だったのが、LEOPARD型の変異はSHP2の酵素活性を著しく低下させる(機能喪失型)のに、なぜ機能獲得型のヌーナン症候群とそっくりな症状になるのか、という点です[3]。
その答えは、LEOPARD型変異が「酵素活性を弱める」と同時に「自己阻害も緩める」二重の作用を持つことにあります[3]。安全装置が外れた変異型SHP2は開いた状態になりやすく、足場タンパク質(GAB1やIRS1など)に正常型よりはるかに長く結びついたままになります。リン酸を外す速度自体は遅くても、シグナル複合体の中継点として居座り続けるため、結果的に下流のRAS-ERKへの入力が長引き、持続的な活性化を生むのです。これは「ドミナントネガティブ」的な、いわば「機能獲得に似た表現型」です[3]。
なお、日本ではヌーナン症候群は指定難病195として医療費助成の対象になっています[11]。ただし広範な遺伝子を調べても、臨床的にヌーナン症候群が疑われる患者さんの約40%では既知の遺伝子に変異が見つからないことも知られており、検査で変異が出ないことが診断の否定を意味するわけではありません[11]。

5. もう一つの顔:メタコンドロマトーシス
🔍 関連記事:メタコンドロマトーシス
PTPN11の機能喪失型のうち、片方のアレルだけが働かなくなるタイプは、骨の表面に良性の骨軟骨腫を、長い骨の端や股関節の軟骨内部に内軟骨腫を多発させるメタコンドロマトーシスの原因になります[9]。興味深いことに、この腫瘍は小児期に現れ、多くは成長とともに自然に小さくなる傾向があります[9]。同じPTPN11でも、ヌーナン症候群とは違う臓器・違うメカニズムで病気を起こす好例です。
6. がん原遺伝子としてのPTPN11:体細胞変異とがん
🔍 関連記事:若年性骨髄単球性白血病(JMML)
💡 用語解説:生殖細胞系列変異 と 体細胞変異
生殖細胞系列変異は、精子・卵子の段階から持っていて全身の細胞に共有される変異で、ヌーナン症候群などはこのタイプです。一方体細胞変異は、生まれた後に特定の細胞だけに後天的に生じる変異で、多くのがんがこれにあたります。同じPTPN11でも、前者は「先天性疾患」、後者は「がん」という、まったく違う顔を見せます。
後天的に骨髄の細胞に生じる体細胞性の機能獲得型変異は、小児に多い侵襲性の高い血液がん「若年性骨髄単球性白血病(JMML)」のリスクを大きく高めます。JMML患者さんの約35%でPTPN11の体細胞変異が認められ、最多のドライバー(引き金)です[7]。さらにPTPN11の体細胞変異は、慢性骨髄単球性白血病・骨髄異形成症候群・急性骨髄性白血病・急性リンパ性白血病など多様な血液疾患や、肺・大腸・脳・甲状腺・悪性黒色腫など一部の固形がんにも低頻度ながら関与します[7]。
ヌーナン症候群の乳児がまれに示すJMML様の症状は、多くが一過性の経過をたどり自然に落ち着くことが多く、純粋な体細胞変異によるJMMLとは区別して管理されます。
SHP2はがん細胞の増殖を後押しするだけでなく、前述したPD-1経路を通じて腫瘍に対する免疫を抑える働きも持つため、製薬の世界で最も重要な創薬標的の一つになっています[7]。研究のきっかけとなったのが、SHP2を不活性な「閉じた」かたちに固定するアロステリック阻害剤「SHP099」です[13]。ただしSHP099はあくまで研究用のツール化合物で、実際に臨床試験へ進んでいるのはTNO155など、その発展形にあたる複数のアロステリック阻害薬です[8]。
💡 用語解説:アロステリック阻害剤
酵素の「働く場所(活性中心)」を直接ふさぐのではなく、別の場所に結合してタンパク質全体のかたちを変え、間接的に働きを止める薬です。SHP099は、SHP2を自己阻害された「閉じた」状態に固定することで活性を抑えます。一方、E76Kという強力な変異はこの薬が結合するポケットそのものを消してしまうため、阻害薬が効きにくくなる耐性の原因になることも分かっています[13]。
7. 周産期医学とNIPT:PTPN11を出生前に調べる
🔍 関連記事:ヌーナン症候群とNIPT/父親の高齢化と新生突然変異/COATE法とは
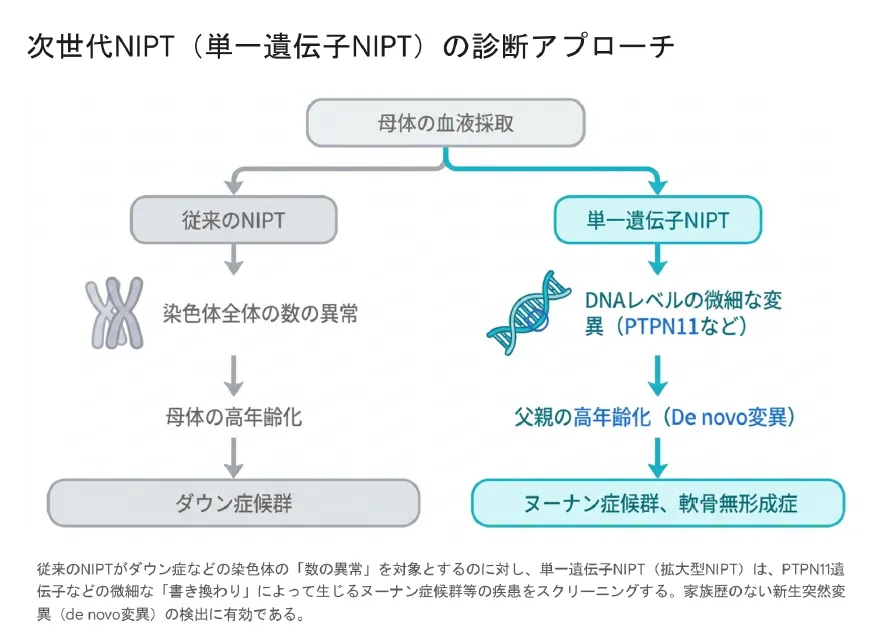
従来のNIPT(新型出生前診断)は、ダウン症候群(21トリソミー)などの「染色体の数の異常」を調べる検査でした。一方、生後に重い障害をもたらす病気の多くは、染色体の数ではなく、DNAの細かな1文字の書き換えで生じる単一遺伝子疾患です[12]。PTPN11によるヌーナン症候群もこのタイプにあたります。
💡 用語解説:新生突然変異(de novo変異)
両親のどちらも持っていないのに、子どもで初めて生じる変異のことです。ヌーナン症候群などのRAS病の多くはこのタイプで、家族歴がない症例が大半を占めます。「家族に同じ病気の人がいないから大丈夫」とは言い切れないのはこのためです。なお、こうした変異は父親の年齢が上がるほど増えやすい傾向が知られています。
最新の次世代シークエンシングの進歩により、母体の血液中にわずかに混じる胎児由来のDNAを高い深さで解析し、PTPN11などの病的変異を採血だけで非侵襲的に調べる「単一遺伝子NIPT」が実用化されています[12]。超音波検査で胎児後頸部の浮腫(NT肥厚)や胸水といったヌーナン症候群を示唆する所見があるハイリスクの妊娠では、確定診断の前に方針を早く決める助けになることが報告されています。ただし精度は超音波所見の有無など検査前の確率に左右されるため、結果の意味づけは遺伝カウンセリングで丁寧に確認することが大切です[12]。
💡 用語解説:胎児フラクション
母体の血液中に含まれる全DNAのうち、胎児(胎盤)由来のDNAが占める割合のことです。この割合が低いとNIPTの精度が下がり、再検査や「判定保留」につながることがあります。母体のBMIが高い場合などに低くなりやすいため、検査時期の調整や事前の説明が重要になります。
ミネルバクリニックでは、PTPN11を含む単一遺伝子疾患をカバーするNIPTとしてダイヤモンドプラン(56遺伝子・30以上の疾患に対応、陽性的中率99.9%超)やインペリアルプランをご用意しています。万一の陽性時には、互助会(8,000円)により羊水検査費用が全額補助されます。検査の詳細や流れはNIPTトップページと互助会のご案内をご覧ください。
8. 遺伝子検査と遺伝カウンセリング
🔍 関連記事:遺伝カウンセリングとは/臨床遺伝専門医とは
PTPN11関連疾患を調べる検査は、「出生前」と「出生後」で目的も方法も異なります。両者を分けて理解することが大切です。
PTPN11関連疾患の多くは新生突然変異(de novo変異)で生じますが、常染色体顕性(優性)遺伝のため、患者さん本人からお子さんへ受け継がれる確率は理論上50%です。確定診断後の遺伝カウンセリングでは、こうした遺伝のしかたと再発リスク、合併症の長期的な見通し、出生前診断の選択肢などを、ご家族の価値観に沿って一緒に整理していきます。検査を受けるかどうか、結果をどう受け止めるかは、つねにご家族が主体です。臨床遺伝専門医は、特定の選択を勧めるのではなく、中立な情報提供者として伴走する役割を担います。
よくある質問(FAQ)
🏥 PTPN11・遺伝子診断のご相談
ヌーナン症候群・LEOPARD症候群などPTPN11に関わる
遺伝子検査・出生前診断・遺伝カウンセリングは
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
参考文献
- [1] PTPN11 gene. MedlinePlus Genetics, U.S. National Library of Medicine. [MedlinePlus]
- [2] Regulation and activity of the phosphatase SHP2: SH2 domains, dephosphorylation activity, and beyond. PMC. [PMC12905495]
- [3] Molecular Basis of Gain-of-Function LEOPARD Syndrome-Associated SHP2 Mutations. Biochemistry. [ACS Biochemistry]
- [4] Single Ion Pair Is Essential for Stabilizing SHP2’s Open Conformation. Biochemistry. [ACS Biochemistry]
- [5] SHP-2 in Lymphocytes’ Cytokine and Inhibitory Receptor Signaling. Frontiers in Immunology. 2019. [Frontiers]
- [6] Hematopoietic Colony Formation from Human Cord Blood Myeloid Progenitor Cells Depends on SHP2 Phosphatase Function. PMC. [PMC3585482]
- [7] Complex Roles of PTPN11/SHP2 in Carcinogenesis and Prospect of Targeting SHP2 in Cancer Therapy. PMC. [PMC11824402]
- [8] Strategies to overcome drug resistance using SHP2 inhibitors. PMC. [PMC8727779]
- [9] Loss-of-Function Mutations in PTPN11 Cause Metachondromatosis, but Not Ollier Disease or Maffucci Syndrome. PMC. [PMC3077396]
- [10] Noonan syndrome with multiple lentigines. MedlinePlus Genetics, U.S. National Library of Medicine. [MedlinePlus]
- [11] ヌーナン症候群(指定難病195). 難病情報センター. [難病情報センター]
- [12] Routine Prenatal cfDNA Screening for Autosomal Dominant Single-Gene Conditions. Clinical Chemistry. 2025. [Clinical Chemistry]
- [13] A Target Mutation that Renders a Cancer Drug Ineffective (SHP2 E76K / SHP099). BioCAT, Advanced Photon Source. [BioCAT/APS]






