目次

DFNB22(常染色体劣性非症候性難聴22)は、内耳にあるOTOA遺伝子の変異によって、音を届けるための「支え」がうまく働かなくなる遺伝性の感音難聴です。音を感じる有毛細胞や聴神経そのものは健康なのに、内耳の蓋膜(がいまく)という膜がずれてしまい、音の振動が神経に伝わりにくくなります。真ん中の高さの音(中音域)が主に聞こえにくくなるのが特徴で、補聴器や人工内耳の効果が高く、近年は根本治療となりうる遺伝子治療の有望な候補としても注目されています。この記事では、原因・症状・診断・治療の最新知見を遺伝専門医の視点でわかりやすく解説します。
Q. DFNB22とはどんな病気ですか?まず結論だけ知りたいです
A. OTOA遺伝子の両方のコピーに変異が起こり、内耳の「オトアンコリン」というタンパク質が働かなくなることで生じる、生まれつきの感音難聴です。蓋膜がラセン縁(内耳の土台)から外れてしまい、音の振動が神経に届きにくくなります。真ん中の音域が主に障害される「中音域難聴」が特徴で、有毛細胞や聴神経は保たれるため、補聴器や人工内耳で良好な聞こえが得られやすい難聴です。
- ➤原因 → 16番染色体(16p12.2)にあるOTOA遺伝子の、両アレルの変異
- ➤病態 → 蓋膜がラセン縁から剥離し、音を伝える「ずれ(剪断流)」が消える
- ➤聞こえの特徴 → 中音域が主に落ちる「クッキーバイト型」の聴力
- ➤診断の難所 → 偽遺伝子OTOAP1とよく似ているため、検査で見逃されやすい
- ➤治療 → 補聴器・人工内耳が良好。単一AAVによる遺伝子治療の有望候補
1. DFNB22とは?ひとことで言うと「音を届ける支えが外れる病気」
DFNB22は、内耳のOTOA遺伝子の変異によって起こる、生まれつきの感音難聴(内耳から聴神経のレベルで生じる難聴)です[1]。「DFNB」は劣性(潜性)遺伝の非症候性難聴を意味する記号で、その22番目に登録された型という意味で「DFNB22」と呼ばれます。「非症候性」とは、難聴以外に目立った合併症を伴わないという意味で、知的発達や見た目、体の他の臓器には基本的に影響が及びません。
「DFNB」の「DF」はdeafness(難聴)を、「N」は非症候性を、「B」は劣性(潜性)遺伝を表し、末尾の番号は発見・登録された順番を示します。つまりDFNB22という名前そのものが、「劣性遺伝で、難聴だけがみられるタイプの、22番目に報告された難聴」という情報を含んでいます。原因遺伝子がまだ特定されていない段階では、この記号だけが手がかりになることもあり、数多くの遺伝性難聴を整理するための共通言語として使われています。
この病気を理解するうえで最も大切なのは、「音を感じ取る細胞(有毛細胞)や聴神経そのものは壊れていない」という点です。壊れているのは、その細胞に音の振動を「届ける」ための力学的な仕組みのほうです。内耳の蓋膜という膜が本来の位置からずれてしまい、音のエネルギーが有毛細胞に伝わらなくなる——いわば「マイクは正常なのに、音源とマイクをつなぐ配線が外れている」ような状態が、DFNB22の本質です[3]。この特徴が、後述する良好な治療反応や遺伝子治療への期待につながっていきます。
💡 用語解説:感音難聴(かんおんなんちょう)とは
耳は「外耳・中耳・内耳」の順に音を伝えます。このうち内耳(蝸牛)や聴神経のレベルで生じる難聴を感音難聴と呼びます。鼓膜や耳小骨など「音を伝える部分」の障害である伝音難聴とは区別されます。感音難聴は音が小さく聞こえるだけでなく、音の輪郭がぼやけて「言葉の聞き取りにくさ」につながりやすいのが特徴です。DFNB22は感音難聴に分類されますが、その中でも「音を感じる細胞は健康で、届ける仕組みだけが壊れる」という、やや特殊なタイプにあたります。
遺伝性難聴の中でDFNB22は決して頻度の高い型ではありませんが、日本人の劣性遺伝性難聴を対象とした大規模な研究では、OTOA遺伝子の変化が全体のおよそ0.3%を占めると報告されています[5]。数字としては小さく見えますが、後述するように「見逃されやすい」性質があるため、実際の頻度はもう少し高い可能性も指摘されています。DFNB22は、遺伝性難聴という大きな地図の中で、原因と仕組みが分子レベルまで解明され、治療の展望まで見えてきた、いわば「研究が進んだ好例」といえる疾患です。
2. 原因遺伝子OTOAとオトアンコリンの働き
🔍 関連記事:OTOA遺伝子の詳細/16p12.2という染色体領域
DFNB22の原因となるOTOA遺伝子は、16番染色体の短い腕(16p12.2)にあり、内耳だけで働く特別なタンパク質「オトアンコリン(Otoancorin)」の設計図です[1]。オトアンコリンは、内耳の「ゲル状の膜」と「その下にある支持細胞の表面」との境目にだけ存在する、接着剤のような役割のタンパク質です[2]。蝸牛では蓋膜を、前庭では耳石膜やクプラ(半規管の感覚を担うゼリー状構造)を、それぞれ細胞表面につなぎとめています。
聞こえに直接関わるのは、蝸牛での働きです。蓋膜の内側の端は、蝸牛の軸に近い「ラセン縁(らせんえん)」という土台に固定されています。この固定を担っているのが、ラセン縁の表面にあるインターデンタル細胞に発現したオトアンコリンです[2][10]。オトアンコリンが失われると、この「蓋膜とラセン縁のつなぎ目」が外れ、蓋膜が土台から浮き上がってしまいます。
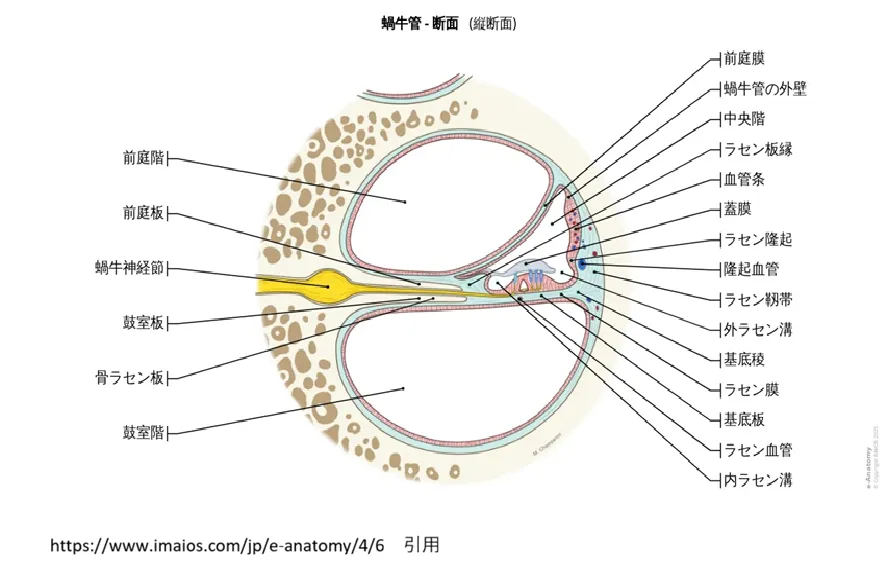
蝸牛管(かぎゅうかん)の断面図。中央階・蓋膜・ラセン板縁(=本文の「ラセン縁」)などの位置関係を示しています。出典:IMAIOS e-Anatomy(https://www.imaios.com/jp/e-anatomy/4/6)より引用
💡 図の見方:「ラセン縁」はどこ?
本文で「ラセン縁」と呼んでいる構造は、この図では「ラセン板縁」と表記されています。どちらも同じ構造(英語でspiral limbus)の別名です。図の右側に並ぶラベルのうち、「中央階」の1つ下・「血管条」の1つ上にある「ラセン板縁」の引き出し線を、内側へたどってください。
位置としては、中央階(蝸牛管の内腔)の内側の底にあり、骨ラセン板の上に乗った、こんもりとした盛り上がりの部分です。このすぐ外側(有毛細胞の側)から蓋膜(がいまく)が伸び出しており、蓋膜の内側の端が、このラセン縁に固定されます。その固定を担うのがオトアンコリンで、DFNB22ではこの接着が外れてしまいます。
内側から外側への並びで覚えると分かりやすく、ラセン板縁(=ラセン縁)→ 内ラセン溝 → コルチ器(有毛細胞)→ 基底膜の順に位置しています。
で音が伝わるしくみ%3C/text%3E%3Ctext x='400' y='56' text-anchor='middle' font-size='13' fill='%237f8c8d'%3E蓋膜がラセン縁にオトアンコリンで固定され、支点となって有毛細胞を刺激します%3C/text%3E%3Crect x='115' y='350' width='570' height='12' rx='6' fill='%23e8eef3' stroke='%23b0bcc6' stroke-width='1.5'/%3E%3Ctext x='400' y='384' text-anchor='middle' font-size='12' fill='%237f8c8d'%3E基底膜(音の振動を受ける膜)%3C/text%3E%3Cpath d='M120 352 Q140 212 195 206 Q246 213 252 352 Z' fill='%23d6eefc' stroke='%233498db' stroke-width='2'/%3E%3Ctext x='184' y='300' text-anchor='middle' font-size='13' font-weight='bold' fill='%232471a3'%3Eラセン縁%3C/text%3E%3Ctext x='184' y='317' text-anchor='middle' font-size='10' fill='%232471a3'%3E(インターデンタル細胞)%3C/text%3E%3Cpath d='M208 216 Q420 198 632 220 Q420 242 208 234 Z' fill='%23fdebd0' stroke='%23e0a800' stroke-width='2'/%3E%3Ctext x='438' y='194' text-anchor='middle' font-size='13' font-weight='bold' fill='%239a6a00'%3E蓋膜(がいまく)%3C/text%3E%3Ccircle cx='214' cy='222' r='5' fill='%23e74c3c'/%3E%3Ccircle cx='209' cy='230' r='5' fill='%23e74c3c'/%3E%3Ccircle cx='219' cy='231' r='5' fill='%23e74c3c'/%3E%3Cline x1='150' y1='256' x2='210' y2='226' stroke='%23e74c3c' stroke-width='1.5'/%3E%3Ctext x='56' y='248' font-size='12' font-weight='bold' fill='%23c0392b'%3Eオトアンコリンが%3C/text%3E%3Ctext x='56' y='264' font-size='12' font-weight='bold' fill='%23c0392b'%3E蓋膜を固定%3C/text%3E%3Cline x1='384' y1='262' x2='384' y2='248' stroke='%231e8449' stroke-width='1.5'/%3E%3Cline x1='391' y1='262' x2='391' y2='246' stroke='%231e8449' stroke-width='1.5'/%3E%3Cline x1='398' y1='262' x2='398' y2='248' stroke='%231e8449' stroke-width='1.5'/%3E%3Crect x='380' y='262' width='26' height='88' rx='6' fill='%23d5f5e3' stroke='%2327ae60' stroke-width='2'/%3E%3Ctext x='393' y='407' text-anchor='middle' font-size='11' fill='%231e8449'%3E内有毛細胞%3C/text%3E%3Ctext x='393' y='421' text-anchor='middle' font-size='11' fill='%231e8449'%3E(IHC)%3C/text%3E%3Cline x1='473' y1='262' x2='473' y2='248' stroke='%237d3c98' stroke-width='1.5'/%3E%3Cline x1='480' y1='262' x2='480' y2='246' stroke='%237d3c98' stroke-width='1.5'/%3E%3Cline x1='487' y1='262' x2='487' y2='248' stroke='%237d3c98' stroke-width='1.5'/%3E%3Crect x='470' y='262' width='22' height='88' rx='6' fill='%23e8daef' stroke='%238e44ad' stroke-width='2'/%3E%3Cline x1='505' y1='262' x2='505' y2='248' stroke='%237d3c98' stroke-width='1.5'/%3E%3Cline x1='512' y1='262' x2='512' y2='246' stroke='%237d3c98' stroke-width='1.5'/%3E%3Cline x1='519' y1='262' x2='519' y2='248' stroke='%237d3c98' stroke-width='1.5'/%3E%3Crect x='502' y='262' width='22' height='88' rx='6' fill='%23e8daef' stroke='%238e44ad' stroke-width='2'/%3E%3Cline x1='537' y1='262' x2='537' y2='248' stroke='%237d3c98' stroke-width='1.5'/%3E%3Cline x1='544' y1='262' x2='544' y2='246' stroke='%237d3c98' stroke-width='1.5'/%3E%3Cline x1='551' y1='262' x2='551' y2='248' stroke='%237d3c98' stroke-width='1.5'/%3E%3Crect x='534' y='262' width='22' height='88' rx='6' fill='%23e8daef' stroke='%238e44ad' stroke-width='2'/%3E%3Ctext x='524' y='407' text-anchor='middle' font-size='11' fill='%237d3c98'%3E外有毛細胞%3C/text%3E%3Ctext x='524' y='421' text-anchor='middle' font-size='11' fill='%237d3c98'%3E(OHC)%3C/text%3E%3Cline x1='362' y1='252' x2='598' y2='252' stroke='%2316a085' stroke-width='2'/%3E%3Cpolygon points='370,247 370,257 358,252' fill='%2316a085'/%3E%3Cpolygon points='590,247 590,257 602,252' fill='%2316a085'/%3E%3Ctext x='645' y='248' font-size='11' font-weight='bold' fill='%23138d75'%3E剪断流(ずれ)で%3C/text%3E%3Ctext x='645' y='264' font-size='11' font-weight='bold' fill='%23138d75'%3E有毛束が曲がる%3C/text%3E%3Cline x1='624' y1='372' x2='624' y2='404' stroke='%233498db' stroke-width='2'/%3E%3Cpolygon points='618,378 630,378 624,368' fill='%233498db'/%3E%3Cpolygon points='618,398 630,398 624,408' fill='%233498db'/%3E%3Ctext x='624' y='424' text-anchor='middle' font-size='11' fill='%232471a3'%3E音による振動%3C/text%3E%3Ctext x='400' y='462' text-anchor='middle' font-size='12' fill='%23555'%3E固定された蓋膜が「支点」となり、振動のたびに有毛束が曲がって電気信号が生まれます%3C/text%3E%3C/svg%3E)
蓋膜はラセン縁にオトアンコリンで固定され、音の振動のたびに有毛束を曲げる「支点」として働きます。この固定が音を届ける前提条件です。
GPIアンカーという「膜へのつなぎ留め」の重要性
オトアンコリンは、細胞膜に「GPIアンカー」という脂質のいかりで係留されるタイプのタンパク質です[4]。韓国の感音難聴家系で見つかったフレームシフト変異(p.Gln589ArgfsX55)を用いた研究では、589番目より後ろのC末端を失った変異型オトアンコリンは、GPIアンカーがうまく付かず、細胞表面にとどまれずに周囲へ流れ出てしまうことが示されました[4]。つまり、このC末端は「膜につなぎ留めるための持ち手」であり、そこを失うとオトアンコリンは接着剤として機能できなくなります。DFNB22の本質が「オトアンコリンが定位置に留まれないこと」にあると分かる、重要な実験的証拠です。
💡 用語解説:GPIアンカーとは
タンパク質を細胞膜の表面につなぎ留める「脂質でできたいかり(アンカー)」の一種です。一部のタンパク質は、このGPIアンカーによって細胞の外側表面に固定され、そこで初めて接着や信号のやりとりといった仕事ができます。オトアンコリンもこのタイプで、GPIアンカーが正しく付くことで、蓋膜と細胞をつなぐ「接着ポイント」として働きます。設計図であるOTOA遺伝子の後半が壊れると、この持ち手が作られず、タンパク質は流れ去ってしまうのです。
3. なぜ難聴になるのか:モデルマウスが解き明かした仕組み
「オトアンコリンが失われると、なぜ音が聞こえなくなるのか」——その正確な仕組みは、オトアンコリンを欠損させたモデルマウスの内耳を詳しく調べた研究で明らかになりました[3]。健康な蝸牛では、蓋膜はラセン縁にしっかり固定され、音の振動が伝わるたびに、蓋膜と有毛細胞の間に「放射状のずれ(剪断流)」が生まれます。このずれが有毛細胞のセンサー(有毛束)を曲げ、機械的な振動を電気信号へ変換します。
ところがオトアンコリン欠損マウスでは、蓋膜がラセン縁の表面から完全に「剥離」して浮いた状態になります[3]。固定という「支点」を失った蓋膜は、音の振動に合わせてコルチ器と同じ位相で滑ってしまい、本来生じるはずの剪断流が消えてしまいます。その結果、とりわけ音の入り口である内有毛細胞(IHC)のセンサーが曲がらなくなり、聴神経へ音の信号が届かなくなります。研究では、8〜70 kHzという広い音域で、神経へ伝わる信号のしきい値が35〜55 dB上昇したことが確認されています[3]。
%3C/text%3E%3Cellipse cx='110' cy='180' rx='34' ry='24' fill='%23d6eefc' stroke='%233498db' stroke-width='2'/%3E%3Ctext x='110' y='184' text-anchor='middle' font-size='10' fill='%232471a3'%3Eラセン縁%3C/text%3E%3Cpath d='M140 165 Q250 150 340 170 Q250 185 140 190 Z' fill='%23f9e79f' stroke='%23e0a800' stroke-width='2'/%3E%3Ctext x='250' y='150' text-anchor='middle' font-size='11' fill='%239a6a00'%3E蓋膜(固定)%3C/text%3E%3Ccircle cx='142' cy='177' r='5' fill='%23e74c3c'/%3E%3Ctext x='250' y='215' text-anchor='middle' font-size='11' font-weight='bold' fill='%231e8449'%3E剪断流あり → 有毛束が曲がる%3C/text%3E%3Crect x='150' y='230' width='200' height='34' rx='4' fill='%23eafaf1' stroke='%2327ae60' stroke-width='2'/%3E%3Ctext x='250' y='252' text-anchor='middle' font-size='13' font-weight='bold' fill='%231e8449'%3E音が脳へ伝わる(聞こえる)%3C/text%3E%3Cellipse cx='510' cy='180' rx='34' ry='24' fill='%23fdecea' stroke='%23e74c3c' stroke-width='2'/%3E%3Ctext x='510' y='184' text-anchor='middle' font-size='10' fill='%23c0392b'%3Eラセン縁%3C/text%3E%3Cpath d='M545 140 Q655 128 745 145 Q655 162 545 165 Z' fill='%23f9e79f' stroke='%23e0a800' stroke-width='2' stroke-dasharray='4,3'/%3E%3Ctext x='650' y='122' text-anchor='middle' font-size='11' fill='%23c0392b' font-weight='bold'%3E蓋膜が剥離(浮遊)%3C/text%3E%3Ctext x='545' y='188' font-size='16' fill='%23e74c3c'%3E✕%3C/text%3E%3Ctext x='650' y='215' text-anchor='middle' font-size='11' font-weight='bold' fill='%23c0392b'%3E剪断流が消える → 有毛束が曲がらない%3C/text%3E%3Crect x='550' y='230' width='200' height='34' rx='4' fill='%23fdecea' stroke='%23e74c3c' stroke-width='2'/%3E%3Ctext x='650' y='252' text-anchor='middle' font-size='13' font-weight='bold' fill='%23c0392b'%3E音が伝わらない(難聴)%3C/text%3E%3Ctext x='400' y='320' text-anchor='middle' font-size='12' fill='%23555'%3E有毛細胞や聴神経そのものは健康なまま。「音を届ける力学的な仕組み」だけが壊れます%3C/text%3E%3Ctext x='400' y='348' text-anchor='middle' font-size='12' fill='%232471a3' font-weight='bold'%3Eだからこそ、電気で神経を直接刺激する人工内耳が効果を出しやすいのです%3C/text%3E%3C/svg%3E)
左:正常では蓋膜が固定され剪断流が生じ、音が伝わる。右:DFNB22では蓋膜が剥離して剪断流が消え、有毛細胞は健康なのに音が届かない。
重要なのは、外有毛細胞(OHC)の能動的な増幅機能や、その働きを反映する歪成分耳音響放射(DPOAE)などは、ほぼ正常に保たれていた点です[3]。つまり有毛細胞自体は健康で、電気を作り出す力も残っています。壊れているのはあくまで「振動を有毛細胞に伝えるリンク」であり、この「力学的な伝達リンクの破綻」こそがDFNB22の難聴の直接の引き金なのです。
この特徴は、診断のうえでのヒントにもなります。外有毛細胞の働きを反映する耳音響放射が記録できるのに、実際の聞こえや聴性脳幹反応(ABR)は不良という、一見矛盾したように見える所見が得られることがあり、これが「音を感じる細胞ではなく、音を届ける力学的な仕組みに問題がある」ことを示唆する手がかりになる場合があります。
さらに、蓋膜が浮いてしまうと、コルチ器全体の「ブレーキ(制動)」も効きにくくなります。健康な状態ではほとんど起こらない自発耳音響放射(外部の音がないのに耳から音が出る現象)が、オトアンコリン欠損マウスでは約79〜80%と高頻度で観察されました[9]。これは、蓋膜の固定が失われて内耳全体の力学的な安定性が崩れ、外有毛細胞の増幅システムが暴走ぎみに不安定化していることを示しています[9]。

4. 聞こえの特徴:中音域難聴(クッキーバイト型)と進行性
DFNB22の聞こえには、他の遺伝性難聴とは異なる特徴的なパターンがあります。多くの患者さんは両耳の対称的な感音難聴を示し、言葉を覚える前(先天性)から難聴があり、めまいや平衡感覚の異常を伴わない「非症候性」の経過をたどります。聴力検査で最も特徴的なのは、真ん中の高さの音(中音域)が選択的に障害される「中音域難聴」、いわゆる「クッキーバイト型(皿状型)」の聴力像です[5]。
」%3C/text%3E%3Ctext x='360' y='54' text-anchor='middle' font-size='12' fill='%237f8c8d'%3E低い音と高い音は比較的保たれ、真ん中の音域がへこむ聴力パターン%3C/text%3E%3Crect x='90' y='80' width='560' height='260' fill='%23ffffff' stroke='%23d0d7de' stroke-width='1'/%3E%3Cline x1='90' y1='80' x2='90' y2='340' stroke='%2395a5a6' stroke-width='2'/%3E%3Cline x1='90' y1='340' x2='650' y2='340' stroke='%2395a5a6' stroke-width='2'/%3E%3Ctext x='40' y='95' font-size='11' fill='%237f8c8d'%3E0 dB%3C/text%3E%3Ctext x='34' y='210' font-size='11' fill='%237f8c8d'%3E50 dB%3C/text%3E%3Ctext x='34' y='335' font-size='11' fill='%237f8c8d'%3E100 dB%3C/text%3E%3Cline x1='90' y1='210' x2='650' y2='210' stroke='%23eee' stroke-width='1'/%3E%3Ctext x='140' y='360' font-size='11' fill='%237f8c8d'%3E250%3C/text%3E%3Ctext x='250' y='360' font-size='11' fill='%237f8c8d'%3E1k%3C/text%3E%3Ctext x='370' y='360' font-size='11' fill='%237f8c8d'%3E2k%3C/text%3E%3Ctext x='490' y='360' font-size='11' fill='%237f8c8d'%3E4k%3C/text%3E%3Ctext x='610' y='360' font-size='11' fill='%237f8c8d'%3E8k(Hz)%3C/text%3E%3Ctext x='360' y='388' text-anchor='middle' font-size='12' fill='%23555'%3E周波数(低い音 ← → 高い音)%3C/text%3E%3Cpolyline points='140,120 250,150 310,235 370,250 430,225 490,150 610,130' fill='none' stroke='%233498db' stroke-width='4'/%3E%3Ccircle cx='140' cy='120' r='6' fill='%233498db'/%3E%3Ccircle cx='250' cy='150' r='6' fill='%233498db'/%3E%3Ccircle cx='310' cy='235' r='6' fill='%23e74c3c'/%3E%3Ccircle cx='370' cy='250' r='6' fill='%23e74c3c'/%3E%3Ccircle cx='430' cy='225' r='6' fill='%23e74c3c'/%3E%3Ccircle cx='490' cy='150' r='6' fill='%233498db'/%3E%3Ccircle cx='610' cy='130' r='6' fill='%233498db'/%3E%3Ctext x='360' y='300' text-anchor='middle' font-size='12' font-weight='bold' fill='%23c0392b'%3E中音域(1〜4kHz)が最も低下 = 皿状のへこみ%3C/text%3E%3C/svg%3E)
低い音と高い音は比較的保たれ、中音域が最も落ち込む「皿状(クッキーバイト型)」の聴力像。クッキーを一口かじった跡のような形からこの名で呼ばれます。
💡 用語解説:中音域難聴(クッキーバイト型)とは
オージオグラム(聴力を高さ別にグラフにしたもの)で、低い音と高い音は比較的よく聞こえるのに、真ん中あたりの音域だけがへこむように落ち込むパターンです。グラフの形がクッキーを一口かじった跡に似ていることから、こう呼ばれます。多くの遺伝性難聴が「高い音から落ちる」タイプであるのに対し、この中音域中心の低下はDFNB22を疑うヒントになります。蝸牛の中で、中音域を感じる部分の蓋膜が、特にオトアンコリンの支えに依存しているためと考えられています[3]。
日本人の難聴患者さんを対象にした大規模なコピー数変異(後述)のスクリーニングでは、2,262例のうち14例でOTOA関連の変化が同定され、そのすべてがこの中音域難聴のパターンを示したと報告されています[5]。この一貫性は、聴力像そのものが診断の重要な手がかりになることを意味します。
DFNB22では、平衡感覚をつかさどる前庭機能はおおむね保たれ、めまいやふらつきを伴わないことが多いとされています。また「音の高さによって聞こえに差がある」という点も臨床的に重要です。会話でよく使う中音域が落ち込むため、本人は聞き返しの多さや言葉の聞き取りにくさを自覚しやすい一方、低音や高音は比較的保たれるため、周囲が難聴に気づきにくいことがあります。こうした聞こえのパターンを丁寧に把握することが、適切な支援につなげる第一歩になります。
かつてDFNB22は「進行しない安定した難聴」と考えられることが多かったのですが、近年の長期的な追跡調査により、この見方は見直されつつあります。遺伝的背景が判明した大規模な難聴コホートの解析では、OTOA遺伝子に変異を持つお子さんの多くで、10年あたり10 dBを超える割合でゆっくりと難聴が進む「進行性感音難聴」の経過をたどる例が示されました[11]。これは、蓋膜の剥離が長期にわたって内耳の環境に力学的なストレスを与え続け、二次的な変化を招くためと推測されています。したがって、一度診断がついた後も、定期的な聴力の確認が大切になります。
5. 診断の難しさ:偽遺伝子OTOAP1という「そっくりさん」
🔍 関連記事:偽遺伝子とは/コピー数変異(CNV)/ロングリードシーケンス
DFNB22の診断が難しいのは、OTOA遺伝子のすぐ近くに、配列が非常によく似た「偽遺伝子OTOAP1」という“そっくりさん”が存在するためです[6]。この領域は進化の過程で生じた「分節重複」を多く含み、配列が繰り返し重なる複雑な構造をしています。似た配列が並んでいると、細胞分裂の際に位置を取り違えた組み換え(非対立遺伝子相同組換え・NAHR)が起こりやすく、OTOA遺伝子の大きな欠失や重複といったコピー数変異(CNV)が生じやすくなります。
💡 用語解説:偽遺伝子とコピー数変異(CNV)
偽遺伝子とは、本物の遺伝子とよく似た配列を持ちながら、タンパク質を作る働きは失っている“化石のような”配列です。OTOAP1はOTOAの偽遺伝子で、配列がそっくりなために検査を混乱させます。
コピー数変異(CNV)は、遺伝子の一部やまとまった領域が「まるごと欠けたり・増えたり」する変化です。1文字だけ変わる点変異と違い、広い範囲が失われるため、通常の検査では捉え方が異なります。DFNB22ではこのCNVが主要な原因の一つで、両アレルの欠失や、片アレルの欠失+もう片アレルの点変異という組み合わせで起こります[5][8]。
もう一つの重要な原因が、偽遺伝子OTOAP1から本物のOTOAへ配列が写し取られる「遺伝子変換」です。遺伝子変換とは、よく似た配列の間で片方の情報がもう片方へコピーされる現象で、OTOAP1に由来する配列がOTOAのエキソン22に入り込むことで、途中でタンパク質合成を止めてしまう変異(p.Glu787*)が生じ、機能しない短いオトアンコリンが作られます[6]。この遺伝子変換は、従来のナンセンス変異(途中で合成が止まる変異)の検査では、そっくりな配列に読み取りが誤って割り当てられてしまい、見逃されやすいという診断上の落とし穴があります[6]。
こうした「そっくりさん問題」を乗り越えるために、近年は長い塩基配列を一気に読み取るロングリードシーケンス(ナノポア解析など)や、コピー数の増減を捉えるMLPAといった手法が活用されています。実際、ロングリード解析によって、OTOAP1由来の配列がどのようにOTOAへ入り込んだのかが正確に描き出され、変換されるDNA断片の長さが数百塩基対から9 kbpを超えるものまで多様であることも分かってきました[6]。
実際の診断は、多くの場合、新生児聴覚スクリーニングや幼少期の聞こえの気づきをきっかけに始まります。まず耳音響放射(OAE)や聴性脳幹反応(ABR)で難聴の有無と程度を確認し、感音難聴が疑われる場合に遺伝学的検査へと進みます。近年は複数の難聴原因遺伝子を一度に調べる遺伝子パネル検査が主流になっていますが、OTOAに関しては、1文字単位の点変異だけを見る通常の解析に加えて、コピー数変異や遺伝子変換を捉える追加の手法を組み合わせることが、見落としを防ぐうえでとくに重要になります[5][6]。
中音域難聴という聴力パターンと、CNVや遺伝子変換に強い検査を組み合わせること——これがDFNB22を見逃さず捉える鍵になります。
6. 遺伝形式と家族:常染色体劣性(潜性)遺伝と隣接遺伝子欠失
DFNB22は常染色体劣性(潜性)遺伝という形式をとります。私たちは同じ遺伝子を父方・母方から1本ずつ、計2本持っています。常染色体劣性遺伝では、2本とも変異している場合にのみ症状が現れ、1本だけ変異している人(保因者)は難聴を示しません。両親がともにOTOAの保因者である場合、お子さんが難聴になる確率は理論上4分の1(25%)です。血縁関係のあるご夫婦(近親婚)では、同じ変異を共有しやすいため頻度が上がることが知られています[7]。
💡 用語解説:保因者(キャリア)とは
ある遺伝子の変異を1本だけ持っているものの、自分自身は症状が出ない人のことです。常染色体劣性(潜性)遺伝の病気では、健康な人でも誰もが何らかの病気の保因者であることが普通です。保因者どうしのカップルから、一定の確率で症状を持つお子さんが生まれる可能性があります。保因者であること自体は「異常」ではなく、ヒトが持つ多様性の一部として理解することが大切です。
一方で、OTOA周辺の領域は前述のとおり欠失が起こりやすく、欠失の大きさによって病像が変わりうる点に注意が必要です。欠失がOTOAだけにとどまる場合は非症候性の難聴となりますが、欠失がOTOAを越えて隣接する遺伝子まで巻き込むほど大きくなると、いくつかの報告では、難聴に加えて発達の遅れなどの神経発達症状を伴う「症候性」の病像が観察されることがあります[7]。これは、失われる遺伝子の範囲が広がることで、難聴以外の機能にも影響が及ぶ可能性を示しています。ただし、どの遺伝子の欠失が神経発達にどこまで関与するかは症例によって異なり、断定できる段階ではありません。
臨床の現場でこの区別が重要なのは、「難聴が単独のものか、それとも大きな欠失に伴う症候性のものか」によって、その後に必要となる発達のフォローが変わってくるためです。こうした「1つの欠失が複数の遺伝子を同時に巻き込む」病態は隣接遺伝子症候群やゲノム病として整理されており、なお、16番染色体のこの近傍領域は難聴以外の微細欠失症候群も知られる複雑な場所です。だからこそ、単に「原因はOTOA」で終わらせず、欠失の範囲まで丁寧に評価し、遺伝カウンセリングの中でご家族に見通しを共有していくことが求められます。
7. 治療と予後:補聴器・人工内耳の良好な成績と遺伝子治療の展望
🔍 関連記事:遺伝子治療とは/AAVベクター/OTOF遺伝子(DFNB9)
DFNB22は、有毛細胞や聴神経が保たれているという病態から、聞こえを補う治療との相性がとても良い難聴です。中音域が主に障害されるため、軽度〜中等度の段階では、残っている低音域・高音域を活かした周波数調整を施した補聴器が高い効果を示します[11]。難聴が進んで高度〜重度に達した場合には、人工内耳が選択肢となります。人工内耳は、内耳の中で電気刺激によって聴神経を直接刺激する装置です。
臨床研究では、OTOA変異を持つ難聴のお子さんは、人工内耳の手術後の言葉の聞き取りや言語獲得で良好な成績を収めやすいことが示されています[11]。これは、電気刺激を脳へ伝えるための螺旋神経節細胞や聴神経が完全に温存されているためです。したがって、早い段階で遺伝子診断を確立しておくことは、術後の見通しをより正確に予測し、ご家族が納得して選択を進めるうえで、大きな助けになります[14]。
難聴のあるお子さんでは、言葉を獲得する大切な時期に、できるだけ早く適切な聞こえの環境を整えることが、その後の言語やコミュニケーションの発達に大きく影響するとされています[14]。DFNB22のように補聴器や人工内耳との相性が良いタイプでは、早い段階で正確な診断と支援につなげることの意義が、とくに大きいといえます。
内耳遺伝子治療という新しい潮流
近年、遺伝性難聴の分野は大きな転換点を迎えています。2026年4月23日、米国食品医薬品局(FDA)は、OTOF(オトフェリン)遺伝子の変異による難聴に対する遺伝子治療薬「Otarmeni(lunsotogene parvec-cwha)」を承認しました[12]。これは遺伝性難聴に対する世界初の遺伝子治療薬で、内耳へ正常な遺伝子を届けるアプローチが現実の治療になりうることを示した歴史的な出来事です。
⚠️ 補足:OtarmeniはOTOF遺伝子(DFNB9)を対象とした薬であり、DFNB22(OTOA)の治療薬ではありません。ここでは「内耳遺伝子治療が実用化された先行事例」として紹介しています。
この流れの中で、OTOAは遺伝子治療に技術的に適した特徴を持つと考えられています。OTOFは設計図(コード領域)が約6 kbと大きく、1つの運び屋ウイルスに収まらないため、2つのAAVベクターに分けて細胞内で再構成させる複雑な仕組みを必要とします[12]。これに対しOTOAのコード領域は約3.4 kbとコンパクトで、標準的な単一AAVベクターに、必要な調節配列とともにそのまま収めることが可能と考えられます[13]。これは、複数ベクターを使わずに済む、扱いやすい標的であることを意味します。
ただし、実際の臨床応用にはまだ超えるべき課題があります。オトアンコリンは有毛細胞そのものではなく、ラセン縁のインターデンタル細胞などの「支持細胞」で作られるため、これらの細胞に効率よく遺伝子を届ける専用の運び屋(特異的なAAVカプシド)の設計が必要です[13]。また、蓋膜が形成される内耳の発生早期に合わせて治療を行うことが、剥離・変形が起こる前に蓋膜を正しくつなぎ直すために重要と考えられており、投与時期の最適化が今後の研究課題です[3]。現時点でOTOAに対する遺伝子治療はあくまで研究・前臨床段階であり、DFNB22の患者さんが今すぐ使える治療ではないことは、正しく理解しておく必要があります。
8. よくある誤解
誤解①「難聴だから音を感じる細胞が壊れている」
DFNB22では、音を感じる有毛細胞や聴神経は健康で、壊れているのは「音を届ける支え」の部分です。だからこそ、電気で神経を直接刺激する人工内耳との相性が良いと考えられています。
誤解②「一度検査で陰性なら原因ではない」
OTOAはそっくりな偽遺伝子があるため、通常の検査では見逃されやすい特徴があります。中音域難聴のパターンがあれば、CNVや遺伝子変換に強い検査を追加で検討する意義があります。
誤解③「進行しない難聴だから放っておいてよい」
かつては安定型と考えられていましたが、近年はゆっくり進行する例が報告されています。診断後も定期的に聞こえを確認し、変化に応じて対応を見直すことが大切です。
誤解④「遺伝子治療がもう使える」
承認されたのはOTOF(別の遺伝子)に対する薬です。OTOAの遺伝子治療は研究・前臨床段階で、DFNB22の患者さんが今すぐ使える治療ではありません。
9. 遺伝専門医からのメッセージ
DFNB22は、原因遺伝子・分子メカニズム・聴力像・治療反応・将来の展望までが、一本の線でつながって理解できる疾患です。「音を感じる仕組みは無事で、届ける支えだけが外れている」という本質を知ることは、ご家族が過度に不安を抱え込まず、次の選択へ進むための土台になります。診断を確定させ、聞こえの経過を見守りながら、その時々で最適な選択肢を一緒に考えていくことが大切です。
遺伝性難聴は、GJB2遺伝子による最も頻度の高いタイプから、DFNB22のような比較的まれなタイプまで、多くの原因遺伝子が知られています[15]。原因を分子レベルで突き止めることは、見通しの共有・適切な支援・将来の治療への橋渡しという点で、大きな価値を持ちます。気になることがあれば、遺伝の専門家に相談するという選択肢があることを知っておいていただければと思います。
よくある質問(FAQ)
🏥 遺伝性難聴・遺伝子診断のご相談
DFNB22をはじめとする遺伝性難聴の
原因遺伝子の検査・遺伝カウンセリングは
臨床遺伝専門医が在籍するミネルバクリニックにご相談ください。
参考文献
- [1] Autosomal recessive nonsyndromic hearing loss 22 (DFNB22), NCBI MedGen. [NCBI MedGen]
- [2] Otoancorin, an inner ear protein restricted to the interface between the apical surface of sensory epithelia and their overlying acellular gels, is defective in autosomal recessive deafness DFNB22. PNAS. 2002. [PNAS]
- [3] A mouse model for human deafness DFNB22 reveals that hearing impairment is due to a loss of inner hair cell stimulation. PNAS. 2012. [PNAS]
- [4] Clarification of glycosylphosphatidylinositol anchorage of OTOANCORIN and human OTOA variants associated with deafness. PMC. [PMC6467692]
- [5] Mid-Frequency Hearing Loss Is Characteristic Clinical Feature of OTOA-Associated Hearing Loss. Genes (Basel). 2019. [MDPI Genes]
- [6] Molecular characterization of pathogenic OTOA gene conversions in hearing loss patients. PMC. [PMC8750238]
- [7] A 250-kb Microdeletion Identified in Chromosome 16 Is Associated With Non-Syndromic Sensorineural Hearing Loss in a South Indian Consanguineous Family. J Audiol Otol. [J Audiol Otol]
- [8] Compound Heterozygosity for OTOA Truncating Variant and Genomic Rearrangement Cause Autosomal Recessive Sensorineural Hearing Loss in an Italian Family. Audiol Res. [MDPI Audiol Res]
- [9] Increased Spontaneous Otoacoustic Emissions in Mice with a Detached Tectorial Membrane. PMC. [PMC4791414]
- [10] Tectorial membrane: structure, function, and its implications for hearing loss. PMC. [PMC12399869]
- [11] Association of Genetic Diagnoses for Childhood-Onset Hearing Loss With Cochlear Implant Outcomes. PMC. [PMC9857764]
- [12] FDA Approves First-Ever Gene Therapy for Treatment of Genetic Hearing Loss Under National Priority Voucher Program. U.S. FDA. 2026. [FDA]
- [13] Advancements and future prospects of adeno-associated virus-mediated gene therapy for sensorineural hearing loss. Front Neurosci. 2024. [Frontiers]
- [14] The Importance of Early Genetic Diagnostics of Hearing Loss in Children. PMC. [PMC7558651]
- [15] Genetics of non syndromic hearing loss. PMC. [PMC4646903]






