目次

📍 クイックナビゲーション
CMT2O(シャルコー・マリー・トゥース病 軸索型2O)は、DYNC1H1遺伝子の変化によって、神経細胞のなかで荷物を運ぶ「逆行性軸索輸送」がうまく働かなくなる、軸索型の遺伝性末梢神経の病気です。多くは小児期に発症し、足から始まる筋力低下・歩きにくさ・凹足(土踏まずの甲高変形)などがゆっくり進みます。100万人に1人未満とされる、とても希少な疾患です。
Q. CMT2Oとはどのような病気ですか?まず結論だけ知りたいです
A. DYNC1H1遺伝子の変化により、神経細胞のなかの「逆行性軸索輸送」が障害されて起こる、軸索型のシャルコー・マリー・トゥース病(遺伝性末梢神経疾患)です。小児期から足を中心とした筋力低下・歩きにくさ・凹足などが、年単位でゆっくり進みます。末梢神経に症状が限られるタイプでは、知的障害を伴わないことがほとんどです。
- ➤疾患の定義 → OMIM 614228、常染色体顕性(優性)遺伝、有病率100万人に1人未満
- ➤原因 → DYNC1H1遺伝子の変化による、神経の「逆行性軸索輸送」の破綻
- ➤主な症状 → 下肢遠位優位の筋力低下・筋萎縮、運動発達の遅れ、凹足、鶏歩
- ➤スペクトラム → SMALED1から大脳皮質形成異常まで連続する「ダイニノパチー」
- ➤診断・管理 → 神経伝導検査+遺伝子検査で確定、対症療法と多職種連携
1. CMT2Oとは:疾患の定義・疫学・歴史
シャルコー・マリー・トゥース病(CMT)は、手足の末梢神経が少しずつ傷んでいく遺伝性の病気の総称で、遺伝性運動感覚ニューロパチー(HMSN)とも呼ばれます。手足の先(遠位)の筋力低下・筋萎縮・感覚の鈍さ・腱反射の低下・凹足などが特徴です。CMTは電気生理学的な性質によって、神経を包む「さや(髄鞘)」が傷む脱髄型(CMT1)と、神経の本体である軸索が傷む軸索型(CMT2)に大きく分けられます。
この記事で扱うCMT2O(シャルコー・マリー・トゥース病 軸索型2O、OMIM 614228)は、この軸索型(CMT2)に属する常染色体顕性(優性)遺伝のサブタイプです。CMT全体の有病率は10万人あたり約18人と推定されますが、その多くは脱髄型のCMT1で占められます。これに対しCMT2Oは極めて稀で、その有病率は100万人に1人未満と推定されています。主に小児期早期に発症し、下肢遠位部を中心とした筋力低下・筋萎縮、運動発達の遅れ、そして人によって幅のある感覚の障害を特徴とします。
💡 用語解説:常染色体顕性(優性)遺伝
「常染色体」とは、性別を決めるX・Y以外の染色体のことです。「顕性(旧:優性)」とは、ペアになっている2本の遺伝子のうちどちらか1本に変化があるだけで症状が現れる性質を意味します。CMT2Oの場合、DYNC1H1遺伝子の片方に変化があるだけで発症し、子へ受け継がれる確率は理論上50%です。ただし実際には、両親に変化がなく子で初めて生じる「新生突然変異(de novo)」での発症も多く知られています。
CMT2OがDYNC1H1遺伝子と結びついたのは、2011年のWeedonらによるエクソーム解析が出発点です。23人の患者を含む4世代の大家系で、従来の検査では原因が見つからなかったところ、全エクソームシーケンスによってDYNC1H1遺伝子に新しい変化が見つかり、家系内の患者と一致して受け継がれていることが確認されました。この発見によって「CMT2O」という新しいサブタイプが正式に確立されました。
2. 原因遺伝子DYNC1H1と逆行性軸索輸送の破綻
CMT2Oの原因は、14番染色体の長腕(14q32.31)にあるDYNC1H1遺伝子の片方の変化です。この遺伝子は、細胞内で荷物を運ぶ巨大なモータータンパク質「細胞質ダイニン」の中心部品(重鎖)の設計図で、4,646個ものアミノ酸からなる非常に大きなタンパク質をつくります。
💡 用語解説:ダイニン複合体
細胞の中には「微小管」というレールが張りめぐらされており、その上を荷物を積んで動く分子の運搬車が「ダイニン」です。ダイニンはATP(細胞のエネルギー通貨)を使って動き、レールの中心方向(神経細胞では細胞体の側)へ荷物を運びます。DYNC1H1がつくる重鎖はこの運搬車のエンジン部分にあたります。詳しくはダイニンの解説ページもご覧ください。
💡 用語解説:逆行性軸索輸送(ぎゃっこうせいじくさくゆそう)
神経細胞は、細胞体から長い「軸索」を伸ばしています。手足を動かす運動神経では、その長さが1メートル近くにもなります。細胞体から末端へ荷物を送るのが「順行性輸送」、逆に末端から細胞体へ使い終わった部品や成長シグナルを送り返すのが「逆行性輸送」です。この戻り便を担うのがダイニンです。戻り便が滞ると、神経の維持や成長のシグナルが届かなくなり、神経が傷んでいきます。
DYNC1H1遺伝子に変化が起きてアミノ酸が1つ入れ替わると、ダイニンの形がわずかに変わり、運搬車としての働きや、他の部品と組み立てる能力が損なわれます。とくに荷物を運ぶ相棒(ダイナクチンやBICD2)と結びついたときに発揮されるべき「長距離をぐんぐん進むモード」が低下し、長い距離の輸送が大きく障害されます。最も長い神経(=足先を支配する神経)から優先的に傷んでいくのはこのためで、CMTで足の症状が先に出る理由をうまく説明できます。
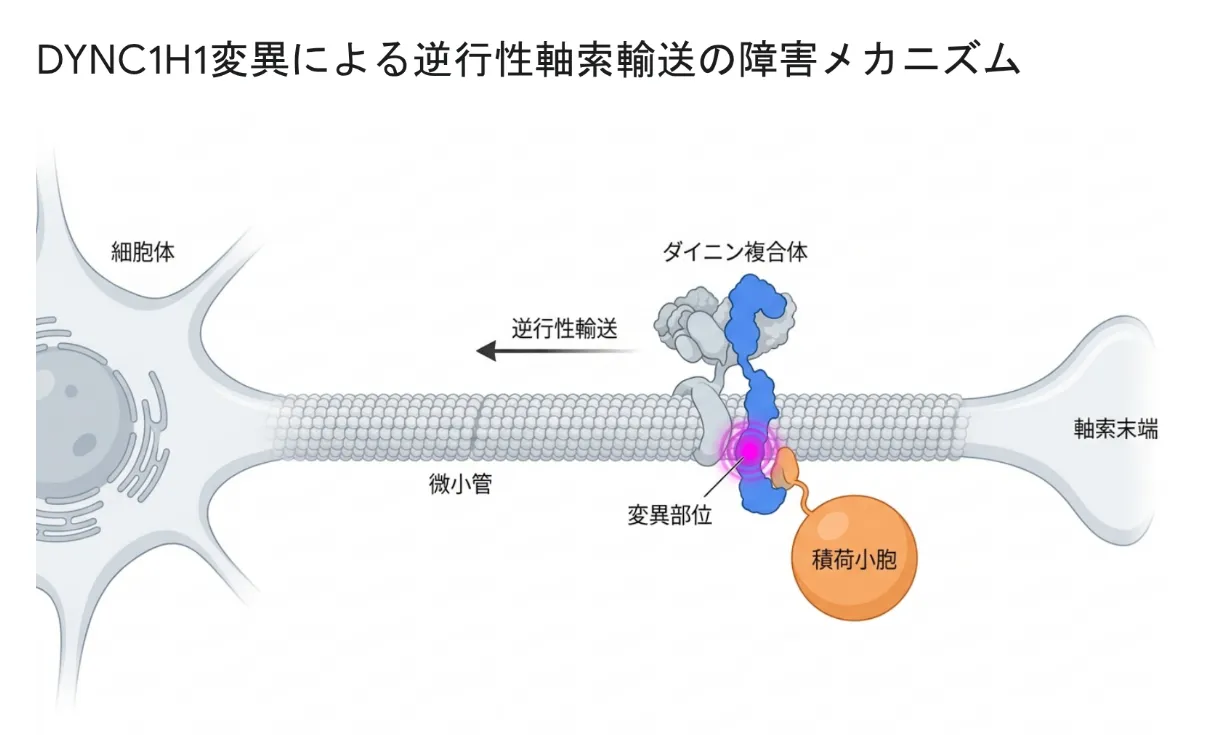
DYNC1H1がつくる細胞質ダイニン重鎖は、軸索に沿ってシナプス小胞などの荷物を細胞体へ運ぶ逆行性輸送を担う。この重鎖の変化によりモーター機能や複合体の組み立てが妨げられ、特に長い軸索をもつ末梢運動神経で軸索の変性が起こる。
💡 用語解説:ミスセンス変異と新生突然変異(de novo)
ミスセンス変異とは、DNAの文字が1つ変わることで、できあがるタンパク質のアミノ酸が別の種類に置き換わるタイプの変化です。タンパク質の形が変わり、はたらきに影響します。CMT2Oの原因変化はこのタイプが中心です。
新生突然変異(de novo)とは、両親には存在せず、精子・卵子がつくられる過程や受精直後に新しく生じた変化のことです。ご両親が健康でも、お子さんで初めて発症することがあります。
なぜ「量が半分」ではなく「邪魔をする」病態なのか
基礎研究の大きな手がかりが、変異マウスの観察です。後肢の異常な姿勢を示す「Loa(Legs at odd angles)」というマウスは、DYNC1H1にあたる遺伝子の変化を持ち、加齢に伴う筋力低下や運動能力の低下を示し、ヒトのCMT2OやSMALEDによく似た像を再現します。重要なのは、この遺伝子を完全に働かなくしただけ(量が半分になるだけ)のマウスでは、このような神経の変性が起こらないという事実です。
💡 用語解説:優性阻害(ドミナントネガティブ)効果
変化した異常なタンパク質が、単に働きを失うだけでなく、正常なタンパク質の働きまで積極的に邪魔してしまう現象です。ダイニンのように複数の部品が集まって機能する複合体では、異常な部品が1つ混じるだけで全体の機能が損なわれることがあります。CMT2Oは、量が足りない「ハプロ不全」ではなく、この優性阻害(または有害な機能獲得)が主な病態と考えられています。
3. 主な症状と自然歴
CMT2O(末梢神経に限られるタイプ)の症状は、主に幼児期から学童期にかけて現れ、きわめてゆっくり進むのが特徴です。症状は、運動神経の障害による筋力低下、感覚神経の障害、そして筋力のアンバランスから二次的に生じる骨格や発達の問題に大きく分けられます。なお、CMT2Oは希少なため、以下の頻度はいずれも限られた症例報告に基づくおおよその目安です。
🦵 運動神経の症状
- 下肢の筋力低下:最大で約95%
- 上肢(手・腕)の筋力低下の合併:約20%
- 運動発達マイルストーンの遅れ:約50%
- 歩行の遅れ・走るのが苦手・転びやすさ
✋ 感覚神経の症状
- 感覚の程度には大きな個人差
- 足を中心とした遠位の感覚鈍麻・振動覚の低下
- 位置覚(固有感覚)の低下で歩行が不安定に
- 成人期に脚の慢性的な疲労感・痛みが出ることも
🦶 骨格・足部の変形
- 凹足(ハイアーチ)・内反尖足(クラブフット)
- 関節拘縮:約10%
- 脊柱側弯症を伴うことも
- 腱反射の低下・消失(下肢中心)
🚶 歩き方・進行
- 動揺性歩行(アヒル歩行):約30%
- つま先が上がりにくい「鶏歩」
- 進行は年単位でとてもゆるやか
- 多くは末梢神経に限られ、知的障害は伴わない
💡 用語解説:凹足(おうそく)と鶏歩(けいほ)
凹足(ハイアーチ)は、土踏まずが通常より高くなる足の変形です。足の筋肉の力のバランスが崩れることで起こり、CMTでよく見られる特徴的な所見です。
鶏歩(けいほ)は、つま先を持ち上げる筋肉が弱くなるため、足先が垂れてつまずかないように、太ももを高く上げて歩く独特の歩き方です。ニワトリの歩き方に似ていることからこう呼ばれます。

4. ダイニノパチー:表現型スペクトラムと鑑別診断
近年の解析で、DYNC1H1遺伝子の変化はCMT2Oという1つの病気にとどまらず、「ダイニノパチー」と総称される非常に幅広い病態を引き起こすことがわかってきました。一方の端には末梢神経だけに症状が限られるタイプ(NMD)があり、もう一方の端には大脳の構造異常を伴う重い神経発達障害(NDD)があります。
末梢神経型(DYNC1H1-NMD)には、CMT2Oのほかに、下肢優位型脊髄性筋萎縮症1型(SMALED1、OMIM 158600)が含まれます。歴史的には別の病気として扱われてきましたが、現在は同じ遺伝子の変化による連続した表現型(アレリック疾患)と理解されています。実際、CMT2Oとして報告された家系で近位の筋力低下が見られたり、SMALED1の患者で遠位の症状や感覚異常が混じったりと、両者の境目はとてもあいまいです。
| 特徴 | DYNC1H1-NMD(CMT2O・SMALED1) | DYNC1H1-NDD(皮質形成異常など) |
|---|---|---|
| 末梢神経の症状 | 運動軸索ニューロパチー、下肢優位の筋萎縮・筋力低下、感覚障害 | 潜在的または軽度(しばしば中枢症状に隠れる) |
| 中枢神経の症状 | 原則として認めない | 知的障害、てんかん、自閉スペクトラム、大脳皮質形成異常 |
| 運動発達・骨格 | 運動発達の遅れ、歩行異常、凹足、関節拘縮 | 全般的発達遅滞、重度の運動機能障害 |
| 変異の主な部位 | テール/二量体化ドメイン(N末端側) | モーター/リンカードメイン(C末端側) |
どの部位の変異がどの症状になるか(遺伝子型-表現型相関)
DYNC1H1タンパク質のどの領域に変化が起きるかによって、症状の重さや中枢神経への影響に傾向があることがわかっています。GeneReviewsなどの標準的な見解では、N末端側のテール(二量体化)ドメインの変化は、主に末梢神経に限られる比較的軽いタイプ(CMT2O・SMALED1)と関連し、C末端側のモータードメインの変化は、大脳皮質形成異常やてんかんを伴う重い中枢神経型(NDD)と関連する傾向があります。実際、CMT2Oの創始変異(His306Arg)は二量体化ドメインに位置しており、この対応関係とよく合致します。
DYNC1H1タンパク質(約4,646アミノ酸)の領域と関連する病型
← N末端 C末端 →
約300〜1140番
末梢神経型
CMT2O・SMALED1
(比較的軽症)
約1374〜1867番
中枢神経型寄り
(発達・行動面)
約1868〜4221番
中枢神経型
皮質形成異常・てんかん
(重症)
同じDYNC1H1の変化でも、領域によって表現型が異なる。CMT2Oは主にテール(二量体化)領域の変化と関連する。
ただしDYNC1H1はとても多面的な遺伝子で、同じ家系・同じ個人のなかでも症状の出方や進み方に大きな幅があります。領域による傾向はあくまで「傾向」であり、最終的な判断には臨床像との総合評価が欠かせません。
他のCMT2サブタイプ・関連疾患との鑑別
CMT2は原因遺伝子によって20以上のサブタイプに分かれ、遺伝的にとても多様です。CMT2Oもこのなかの1つで、原因遺伝子と関わる機能が異なります。また、ダイニンの相棒であるBICD2遺伝子の変化によるSMALED2(OMIM 615290)も、症状が似るため鑑別が必要です。
| サブタイプ | 原因遺伝子 | 関わる機能・病態 |
|---|---|---|
| CMT2A | MFN2 | ミトコンドリアの融合 |
| CMT2B | RAB7A | エンドソーム機能・小胞輸送 |
| CMT2D | GARS | tRNA合成酵素の機能 |
| CMT2E | NEFL | 神経フィラメントの構造・輸送 |
| CMT2O | DYNC1H1 | 逆行性軸索輸送の障害 |
| CMT2P | LRSAM1 | ユビキチンリガーゼ機能 |
5. 診断の進め方(出生後・出生前)
CMT2Oは極めて稀で、ほかの遺伝性末梢神経疾患や脊髄性筋萎縮症、運動発達の遅れと症状が重なるため、正確な診断には複数の角度からのアプローチが欠かせません。ここでは、すでに生まれたお子さん・ご本人を調べる「出生後診断」と、妊娠中の「出生前診断」を分けて説明します。
出生後の診断:電気生理検査+遺伝子検査
まず、神経の働きを調べる神経伝導検査(NCS)と針筋電図(EMG)を行い、脱髄型か軸索型かを見分けます。CMT2Oは軸索型なので、神経の伝わる速さ(NCV)は正常か軽度低下にとどまる一方、軸索が減ることを反映して、神経や筋肉から得られる反応の振幅が低下します。
💡 用語解説:神経伝導検査(NCS)と軸索型・脱髄型
皮膚の上から神経に弱い電気刺激を与え、信号が伝わる速さや大きさを測る検査です。神経のさや(髄鞘)が傷む脱髄型では伝わる速さが大きく落ちます(上肢でおおむね毎秒38メートル未満)。一方、神経の本体が傷む軸索型(CMT2O)では速さは保たれ(毎秒38メートル以上)、反応の大きさ(振幅)が下がるのが特徴です。
臨床と電気生理でCMTが疑われたら、確定のために遺伝子検査を行います。CMT2Oの原因はアミノ酸が1つ入れ替わるミスセンス変異のため、染色体の大きな過不足を見る検査(Gバンド核型分析や染色体マイクロアレイ)では検出できません。文字の並び(塩基配列)を読む次世代シーケンス(NGS)による遺伝子パネル検査や全エクソーム解析(WES)が確定診断の中心です。CMTは原因遺伝子が100種類以上あるため、関連遺伝子をまとめて調べられるパネル検査が効率的です。
当院では、CMTおよび類似疾患の原因遺伝子をまとめて調べるシャルコー・マリー・トゥース病(CMT)遺伝子検査(NGSパネル)を行っており、このパネルにはDYNC1H1も含まれています。検査の一般的な考え方は遺伝子検査とは(メリット・デメリット)もあわせてご覧ください。
出生前の診断:家族内に既知の変化がある場合
ご家族のなかでDYNC1H1の原因となる変化がすでに特定されている場合には、妊娠中に調べる選択肢があります。確定診断としては、羊水検査・絨毛検査による胎児の遺伝子解析が行えます。また、新生突然変異による顕性疾患を妊娠中に調べるNIPTのインペリアルプランでは、検査対象の遺伝子のなかにDYNC1H1が含まれています。なお、NIPTはあくまでスクリーニング(ふるい分け)であり、確定には羊水・絨毛検査が必要です。
CMT2Oは生命予後に大きく影響しないことが多く、重症度の幅も広い疾患です。そのため、出生前に調べることが常にご家族の利益になるとは限りません。検査を受けるかどうかは、十分な情報のもとでご家族が主体的に決めることが大切です。当院は特定の検査をおすすめする立場ではなく、中立的に情報をお伝えします。
6. 治療と長期管理
現時点では、変化したDYNC1H1遺伝子を直したり、ダイニンの働きを直接回復させたりする根本的な治療法は確立されていません。そのため管理の中心は、症状をやわらげ、生活の質(QOL)を最大限に保つための対症療法と支持療法になります。CMT2Oは生命予後に大きく影響しないことが多く、多くの方が日常生活や仕事を続けられます。
🤝 多職種チームによるケア
小児神経科・整形外科・リハビリテーション科・理学療法士、そして臨床遺伝専門医や遺伝カウンセラーが連携します。中枢神経の関与が疑われる場合は、言語聴覚士などの介入も加わります。
🦿 リハビリと装具
関節の動きを保ち、拘縮を防ぐための理学療法を続けます。歩行を安定させ転倒を防ぐために、足底板(インソール)や短下肢装具(AFO)などの使用が役立ちます。
🦴 整形外科的な治療
リハビリで対応が難しい重い凹足・内反尖足、戻らない関節拘縮、進行する脊柱側弯症には、腱を延ばす手術や骨切り術などが検討されることがあります。手術の時期は慎重に判断します。
あわせて知っておきたいのが、CMTでは末梢神経を傷めやすい一部の薬剤(抗がん剤のビンクリスチンなど)に注意が必要という点です。新しい薬を処方されるときには、CMTがあることを必ず主治医に伝えてください。
7. 遺伝カウンセリングの意義
CMT2Oの診断がついたあとは、ご家族への丁寧な遺伝カウンセリングが重要になります。臨床遺伝専門医が、次のような内容を中立的な立場でお伝えします。
- ➤遺伝形式と再発のリスク:常染色体顕性(優性)遺伝のため、患者ご本人がお子さんを持つ場合の遺伝確率は理論上50%です。一方で、ご両親に変化がなくお子さんで初めて生じる新生突然変異(de novo)も多く、その場合ご両親への遺伝は通常認めません。ただし生殖細胞のモザイク(一部の細胞だけに変化がある状態)の可能性は完全には否定できません。
- ➤予後の見通し:末梢神経に限られるタイプでは、生命予後に大きく影響しないことが多く、知的障害も伴わないのが一般的です。この情報は、教育や就労を含めた長期的な見通しを立てるうえで、ご家族にとって大切な手がかりになります。
- ➤家族計画の選択肢:既知の変化が特定されている場合は、絨毛検査・羊水検査による出生前診断や、着床前診断といった選択肢を情報としてお示しします。選ぶかどうかはご家族が決めることです。
- ➤長期フォローと情報の蓄積:希少疾患のため自然歴の情報は限られています。2023年には国際的な患者支援団体(The DYNC1H1 Association)も設立され、患者コホートの形成が進んでいます。医療機関との連携を続けることが重要です。
8. よくある誤解
誤解①「DYNC1H1の変化=必ず重い病気」
DYNC1H1の変化は、末梢神経に限られる比較的軽いCMT2Oから、重い中枢神経症状まで幅があります。変化の場所と臨床像をあわせて評価しなければ、重さは決められません。
誤解②「染色体検査で分かる」
CMT2Oはアミノ酸が入れ替わるミスセンス変異が原因のため、染色体の大きな過不足を見る検査では検出できません。塩基配列を読むシーケンス検査が必要です。
誤解③「両親が健康だから遺伝病ではない」
CMT2Oは新生突然変異(de novo)での発症も多く、ご両親に変化がなくてもお子さんが発症することがあります。「両親が健康だから」という思い込みが診断を遅らせることがあります。
誤解④「CMT2Oは脳性麻痺の一種」
運動発達の遅れや歩行異常から脳性麻痺と誤って捉えられることがあります。CMT2Oは末梢神経の遺伝性疾患であり、電気生理検査と遺伝子検査で区別されます。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 遺伝性末梢神経疾患の診断・遺伝カウンセリングについて
CMT2Oをはじめとする遺伝性末梢神経疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にお問い合わせください。
参考文献
- [1] OMIM #614228. Charcot-Marie-Tooth Disease, Axonal, Type 2O (CMT2O). Johns Hopkins University. [OMIM]
- [2] Scoto M, et al. DYNC1H1-Related Disorders. GeneReviews®. University of Washington, Seattle. [GeneReviews / NBK601997]
- [3] Weedon MN, et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. 2011;89(2):308-312. [PubMed 21820100]
- [4] Orphanet. Autosomal dominant Charcot-Marie-Tooth disease type 2O. ORPHA:284232. [Orphanet]
- [5] Becker LL, Di Donato N, et al. The clinical-phenotype continuum in DYNC1H1-related disorders—genomic profiling and proposal for a novel classification. J Hum Genet. 2020;65(11):1003-1017. [Nature / J Hum Genet]
- [6] Harms MB, et al. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology. 2012;78(22):1714-1720. [PMC3359582]
- [7] Li JT, et al. Expanding the Phenotypic and Genetic Spectrum of Neuromuscular Diseases Caused by DYNC1H1 Mutations. Front Neurol. 2022;13:943324. [PMC9309508]
- [8] NIH GARD / NCATS. Charcot-Marie-Tooth disease axonal type 2O. [NIH GARD]






