目次

線維軟骨発生症1型(FBCG1)は、COL11A1遺伝子の変異によって起こる、100万人に1人未満という超希少な骨系統疾患です。短い手足・狭い胸・ダンベルのように両端がふくらんだ長い骨・洋梨のような形の背骨を特徴とし、かつては生まれてすぐに亡くなる致死的な病気と考えられてきました。しかし近年は、人工呼吸管理などの集学的な治療によって学童期・思春期まで成長し、正常な知能を保つ生存例も報告されるようになっています。
Q. 線維軟骨発生症1型とはどんな病気ですか?まず結論だけ知りたいです
A. COL11A1遺伝子の変異により、骨や軟骨のもとになる「XI型コラーゲン」がうまく作れなくなる超希少な骨系統疾患です。短い手足・狭い胸・特徴的なダンベル状の長い骨を示し、新生児期は狭い胸による呼吸の問題が最大の課題となります。一方で知能の発達には直接影響しないことが知られ、近年は集学的治療による長期生存例も報告されています。
- ➤疾患の定義 → OMIM #228520、有病率100万人に1人未満、世界で約20〜22例の報告
- ➤原因 → COL11A1遺伝子の変異と、XI型コラーゲンの構造異常
- ➤主な症状 → ダンベル状の長管骨・洋梨状の扁平椎・狭い胸・難聴・近視
- ➤鑑別診断 → 致死性骨異形成症など、他の重い骨系統疾患との違いを詳しく解説
- ➤診断・管理 → 次世代シーケンサーによる診断と、集学的治療・成長ホルモン療法の実際
1. 線維軟骨発生症1型とは:疾患の定義と歴史
線維軟骨発生症1型(せんいなんこつはっせいしょう1がた/Fibrochondrogenesis 1、略してFBCG1)は、骨と軟骨の発育が広い範囲で障害される、非常に重い先天性の骨系統疾患です。国際的な遺伝病データベースOMIMには「#228520」として、希少疾患データベースOrphanetにも疾患として登録されています。推定有病率は100万人に1人未満と極めて低く、これまで世界の医学文献で報告されたのは約20〜22例にとどまる超希少疾患です。
独立した病気として最初に報告されたのは1978年です。手足が著しく短いこと(短肢症)、胸郭が狭いことによる重い呼吸不全、そして全身の多くの臓器にわたる特徴的な変化を持つ病気として記載されました。とりわけ長い骨(長管骨)の両端がダンベルのようにふくらむ形、背骨が洋梨のように扁平になる形、そして病名の由来となった軟骨細胞の「線維芽細胞のような変化」が、この病気を見分ける核心となります。
FBCG1は、もともと新生児期に肺の発育が不十分で生命維持が難しい「致死的な病気」として分類されてきました。しかし、全エクソーム解析などの遺伝子解析技術が臨床に広まり、新生児集中治療(NICU)が大きく進歩したことで、この理解は転換期を迎えています。生後早期からの人工呼吸管理や気管切開といった積極的な治療によって、乳児期を乗り越え、学童期や思春期まで成長し、知的発達も正常な生存例が報告されるようになりました。
💡 用語解説:常染色体潜性遺伝(じょうせんしょくたいせんせいいでん)
「常染色体」とは、性別を決めるX・Y染色体以外の染色体のことです。「潜性(劣性)」とは、同じ遺伝子のペア(父由来・母由来の2本)の両方に変異がそろってはじめて症状が現れる遺伝のしかたを意味します。FBCG1は主にこの常染色体潜性遺伝で起こり、両親はそれぞれ変異を1つずつ持つ「保因者(症状のない持ち主)」であることが多いのが特徴です。遺伝のしくみについては 遺伝形式の解説ページ でも詳しく説明しています。
さらに近年、表現型(症状の出方)の幅が広いことがわかってきたことで、従来の「潜性遺伝のみ」という定説を見直す動きも出ています。後で詳しく述べますが、顕性(優性)遺伝で重い骨系統疾患を起こす変異や、保因者にも軽い症状(近視や難聴)が現れる例なども明らかになってきました。
2. 原因遺伝子COL11A1とXI型コラーゲンの役割
FBCG1の根本的な原因は、第1番染色体(1p21.1という場所)にあるCOL11A1遺伝子の変異です。この遺伝子は全部で67のエクソン(タンパク質の設計図となる部分)からなり、「XI型コラーゲン」を作るための重要な材料をコードしています。
💡 用語解説:XI型コラーゲンとは
コラーゲンは、体のさまざまな組織を支える「繊維状のタンパク質」です。そのうちXI型コラーゲンは、主に軟骨・内耳・眼の硝子体(しょうしたい)・結合組織に分布し、組織の強度と形を保つ「足場」として働きます。軟骨の中ではII型コラーゲンなどと協力し、コラーゲンの繊維が育つ最初のきっかけを作り、繊維が太くなりすぎないよう調整する「型枠」の役目を担います。この型枠が壊れると、軟骨の構造が大きく乱れてしまいます。
COL11A1に変異が起こると、XI型コラーゲンの精密な分子同士のネットワークが破綻します。その結果、軟骨や結合組織の構造が大きく無秩序になり、胎児期に骨が正常に作られていくプロセス(内軟骨性骨化)が著しく妨げられます。これが、FBCG1の重い骨格異常と低身長を引き起こす直接のしくみです。
💡 用語解説:内軟骨性骨化(ないなんこつせいこっか)
手足の長い骨などが作られるとき、まず「軟骨の鋳型(いがた)」ができ、それが少しずつ本物の骨に置き換わっていきます。このプロセスを内軟骨性骨化といいます。FBCG1ではこの鋳型となる軟骨の質そのものが乱れているため、骨が正しい形に育たず、特徴的なダンベル状の変形が生じます。
どのような変異が見つかるのか
FBCG1の患者さんの多くは、COL11A1遺伝子に機能喪失型の変異とミスセンス変異を1つずつ持つ「複合ヘテロ接合体」として報告されています。たとえば全エクソーム解析で診断された韓国で初めての生存例では、エクソン45の新規変異 c.3478C>G(p.Pro1160Ala)と、エクソン35の c.2771C>T(p.Pro924Leu)の組み合わせが見つかりました。とくに新規のp.Pro1160Alaは、生物種を超えて高度に保存された領域にあり、タンパク質の構造と機能に重大な影響を与える可能性が高い(病的である可能性が高い)と評価されています。
💡 用語解説:ミスセンス変異と複合ヘテロ接合体
ミスセンス変異とは、DNAの塩基が1つ変わることで、設計図の一部のアミノ酸が別の種類に置き換わるタイプの変異です。タンパク質の形が変わり、機能に影響します。くわしくは ミスセンス変異の解説ページ をご覧ください。
複合ヘテロ接合体とは、父由来と母由来の遺伝子に、それぞれ「別々の変異」を1つずつ持っている状態です。同じ遺伝子の2本がともに正常に働けなくなるため、潜性遺伝の病気を発症します。
例外的な遺伝のしかた:顕性遺伝とモザイク
長らくFBCG1は常染色体潜性遺伝の病気と考えられてきましたが、近年の家系解析で顕性(優性)遺伝で重い骨系統疾患を起こす新しいCOL11A1変異も報告され、病気の複雑さがいっそう明らかになっています。ある家系では、母親が軽い四肢短縮・短い指・強い近視を示し、解析の結果「生殖細胞系列モザイク」であることがわかりました。この母親から変異を受け継いだ2人の子は、強い近視・難聴・四肢短縮・骨幹端の拡大・小さなあご・気管切開を要する気道病変など、重い症状を示しました。完全な致死には至らず、第一子は2年以上、第二子は5か月生存しており、コラーゲンの病気における症状の幅広さをよく表しています。
💡 用語解説:ハプロ不全と保因者の症状
ハプロ不全とは、遺伝子のペアのうち片方が働かなくなり、タンパク質の量が半分になることで症状が出る状態です。大規模な研究では、COL11A1のミスセンス変異を片方だけ持つ親(保因者)に近視が、機能喪失型を持つ親に早期発症の難聴が認められました。これは、COL11A1が軽い遺伝性の難聴や近視の単独の原因にもなりうることを示す重要な知見です。一方、2型(COL11A2が原因)の保因者では視覚・聴覚を含めて正常とされ、両遺伝子の働き方の違いが浮き彫りになっています。
3. 主な症状と全身の特徴
FBCG1の症状は、骨格や結合組織の異常を出発点として、全身の多くの臓器に広がります。臓器系ごとに、代表的な特徴を整理します。
🦴 骨格・体つき
- 著しい低身長と、手足の強い短縮
- 釣鐘のように狭い胸郭(呼吸不全の最大要因)
- 背骨が扁平になる扁平椎、極端に短い首
- 長管骨のダンベル状変形・骨幹端の拡大
👶 顔つき(頭蓋顔面)
- 中顔面の平坦さ・丸顔または三角形の輪郭
- 眼球突出・両眼開離・眼裂斜下
- 小さく短い鼻・前方を向く鼻孔(前傾鼻孔)
- 小さなあご(小顎症)・口蓋裂・二分舌
👁️ 眼・耳(感覚器)
- 強い近視(高度近視)・白内障
- 網膜剥離のリスクが高い
- 感音難聴(生存例ではほぼ全例)
🩺 その他の全身所見
- 手指の屈指症・合指症・関節拘縮
- 大きく前に突き出た腹部・臍帯ヘルニア
- 重症例では胎児水腫(体液貯留)
FBCG1で最も生命に関わるのは、釣鐘状に狭い胸郭と短い肋骨が、胎児期の肺の発育を物理的に妨げることです。これが致死的な呼吸不全を引き起こす最大の要因となります。
画像(X線)と組織でみる特徴
FBCG1のX線像は非常にユニークで、診断の決め手になります。長い骨は極端に短い一方で骨幹端が外側に大きく広がり、骨幹部が短く両端が膨らんだ「ダンベル状」の形をとります。背骨を横から見ると、椎体の後ろ側が低形成で狭くなり、前側が丸くふくらむため、「つまんだような」「洋梨状」の特徴的な形になります。肋骨は短く幅広で、両端が盃のようにへこむカッピングがみられます。
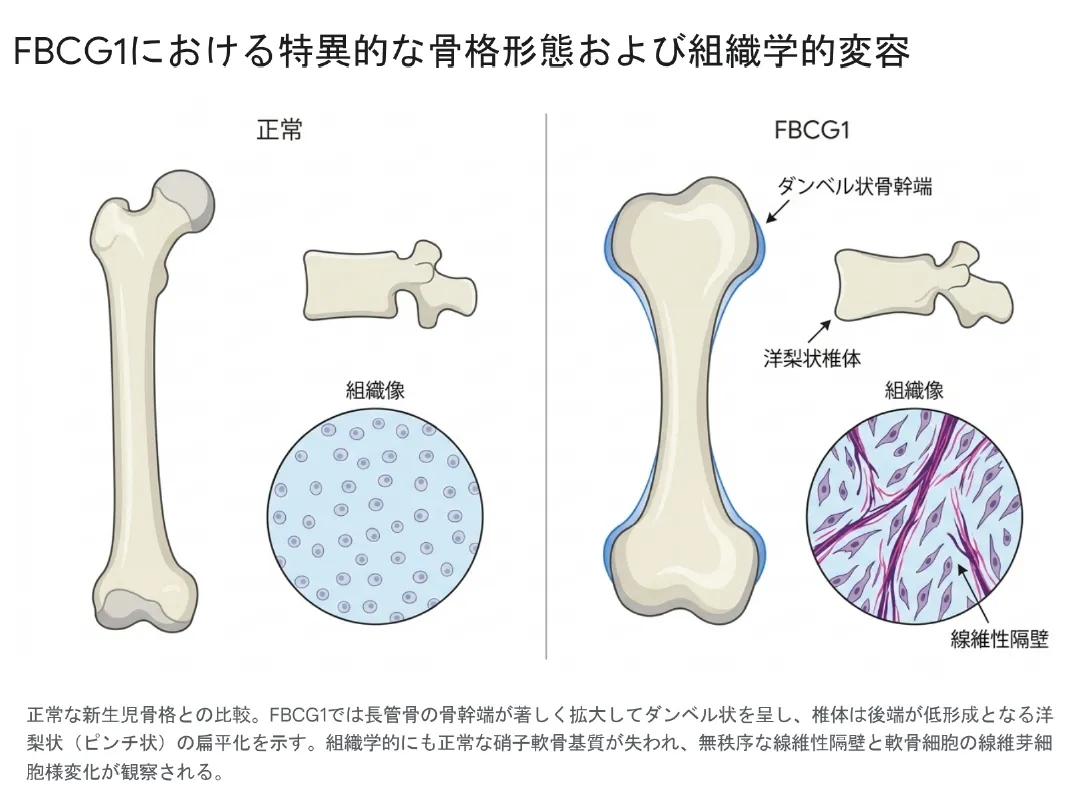
正常な新生児骨格との比較。FBCG1では長管骨の骨幹端が著しく拡大してダンベル状を呈し、椎体は後端が低形成となる洋梨状(ピンチ状)の扁平化を示す。組織学的にも正常な硝子軟骨基質が失われ、無秩序な線維性隔壁と軟骨細胞の線維芽細胞様変化が観察される。
💡 用語解説:線維芽細胞様変化と扁平椎
本来、軟骨の中の細胞(軟骨細胞)は丸い形で規則正しく並んでいます。FBCG1ではこの並びが崩れ、細胞が細長い「線維芽細胞」のような形に変わり、細胞のあいだに無秩序な繊維の仕切り(線維性隔壁)が作られます。病名「線維軟骨発生症」はこの組織の特徴に由来します。
扁平椎(へんぺいつい)とは、背骨の椎体が上下につぶれて平たくなった状態のことです。

4. 鑑別診断:似ている骨系統疾患との違い
「短い手足」と「狭い胸」がみられる場合、致死的または重症の骨系統疾患がいくつも候補に挙がります。FBCG1と他の病気を正確に見分けることは、出生直後の対応方針や、次のお子さんに向けた遺伝カウンセリングのうえで、とても重要な意味を持ちます。
| 疾患名 | 主な原因遺伝子 | FBCG1との見分け方 |
|---|---|---|
| 軟骨無発生症 | TRIP11/SLC26A2/COL2A1 | FBCG1以上に骨化が乏しく、脊椎・骨盤・頭蓋の骨化が著しく欠けます。ダンベル状の長管骨変形は通常みられません。 |
| 致死性骨異形成症 | FGFR3 | 胸郭の狭さは共通ですが、大腿骨が受話器のように湾曲したり、クローバー葉型の頭蓋を伴います。椎体はH型の扁平椎で、FBCG1の洋梨状とは異なります。 |
| 骨形成不全症2型 | COL1A1/COL1A2 | 骨がもろく、周産期に多発骨折を起こします。骨が短く曲がり厚くなる変形や青色強膜が特徴で、軟骨そのものの形成異常とは病態が異なります。 |
| クニースト異形成症 | COL2A1 | ダンベル状の長管骨・近視・難聴など重なりますが、椎体に冠状裂がみられ、軟骨は線維化ではなく「スイスチーズ様」の空洞を示します。一般に致死的ではありません。 |
| 分節異常性異形成症 | HSPG2 | FBCG1が近位部中心の短縮であるのに対し、四肢全体が不均衡に短縮します。背骨の広範な分節異常による重い側弯・後弯を伴います。 |
| 低ホスファターゼ症(周産期致死型) | ALPL | 骨化・石灰化が広範に障害されます。血液検査で血清ALP(アルカリホスファターゼ)が著しく低いことで確定診断されます。 |
このように、似た病気が多いからこそ、X線所見・組織所見・そして遺伝子検査を組み合わせて総合的に判断することが大切になります。
5. 診断の進め方と遺伝子検査
FBCG1の診断では、「お腹の中にいるとき(出生前)」と「生まれたあと(出生後)」で、進め方や使える検査が異なります。ここを分けて理解することが大切です。
出生前:胎児超音波で気づかれることが多い
FBCG1は、妊娠中期から後期の胎児超音波検査で重大な形態異常が指摘され、発見されることが多い病気です。頭の形の異常、胸とお腹のバランスの崩れ(狭い胸郭)、四肢の著しい短縮などが手がかりになります。羊水過多や胎児水腫を伴う場合、産科医はFBCG1を含む骨系統疾患を候補に挙げ、遺伝カウンセリング へとつなげます。
出生前に確定診断を行う場合は、絨毛検査(CVS)や羊水検査で得られた細胞を用いて、原因遺伝子の変異を調べます。費用や手順については 羊水検査・絨毛検査のページ をご参照ください。
確定診断は分子遺伝学的検査で
超音波やX線でFBCG1が疑われたら、確定診断には分子遺伝学的検査(遺伝子検査)が不可欠です。具体的には、ターゲットを絞った次世代シーケンシング(NGS)や、全エクソーム解析(WES)が用いられます。妊娠が進行中の出生前症例では、判断を急ぐため迅速処理が行われることもあります。
💡 用語解説:全エクソーム解析(WES)
遺伝子のうち、タンパク質の設計図となる部分(エクソン)をまとめて網羅的に読み取る検査です。多くの遺伝病の原因がこの領域に集まっているため、原因が1つに絞りきれない場合でも効率よく変異を見つけられます。くわしくは エクソーム解析の解説ページ をご覧ください。
見つかった変異が「病気の原因かどうか」の判定には、ACMG(米国臨床遺伝・ゲノム学会)の基準が用いられます。判定の考え方は バリアント分類の解説ページ でまとめています。多遺伝子パネル検査の活用は、意義不明のバリアント(VUS)の不確実性を減らし、診断の役立ち度を高めることが示されています。
当院では、COL11A1を解析対象に含む遺伝子パネル検査として、結合組織疾患NGSパネル検査 や 低身長遺伝子パネル検査 をご用意しています。また出生前のスクリーニングとしては、COL11A1を含む単一遺伝子をカバーする NIPTダイヤモンドプラン や インペリアルプラン も選択肢として存在します。どの検査が適しているかは、状況に応じて遺伝カウンセリングのなかで一緒に考えていきます。
6. 治療と長期管理
FBCG1には、病気そのものを根本から治す方法はまだありません。しかし、複数の診療科が連携する集学的なケアによって、症状を管理し、生活の質を高めることが可能になってきました。生存例の管理には、小児科・整形外科・呼吸器科・眼科・耳鼻咽喉科・遺伝診療科などのチーム医療が欠かせません。
新生児期:呼吸の確保が最優先
FBCG1がかつて致死的とされた最大の理由は、狭い胸郭による高度の肺低形成と、それに伴う呼吸不全です。生後すぐからの積極的な蘇生と集中治療が、最重症期を乗り越えられるかどうかを左右します。ある生存例では、出生後1か月にわたる持続的な呼吸サポートと、その後数か月の気管切開による管理を経て、長期生存に至りました。一方、強力な集中治療を要さずに自発呼吸を確立できた例もあり、重症度には幅があることがわかっています。
成長ホルモン(GH)療法という選択肢
低身長や成長障害に対する対症療法として、遺伝子組み換えヒト成長ホルモン(GH)治療の有効性を示す報告が積み重なってきています。COL11A1関連の重症骨系統疾患の患者さんにGH治療を行った研究では、治療初年度に年間の成長速度が最大9.1cm/年まで増加し、身長の標準偏差スコア(SDS)がベースラインから+1.5改善したと報告されています。
全エクソーム解析で診断が確定した韓国の男児(生存例)の長期データは、特に示唆に富んでいます。5歳11か月時点で身長105.0cm(-2.09 SDS)だったのが、継続的なモニタリングのもとで成長を続け、12歳時点で145.5cm(-1.00 SDS)に到達しました。下のグラフは、その身長と標準偏差スコアの推移を示したものです。
📈 GH療法を受けた生存例の身長推移
標準偏差スコア(SDS)は -2.09 から -1.00 へと改善しました。SDSは「同年齢の平均からどれだけ離れているか」を示す指標で、0に近いほど平均的な身長に近いことを意味します。
この男児は骨年齢が実年齢より2年先行し、正常な思春期発達の兆候を示しました。そして特筆すべきは、中枢神経の発達に異常がなく、正常な知能が確認されている点です。これは、FBCG1が認知機能や脳の発達そのものには直接の悪影響を及ぼさないことを強く示す、重要な臨床的証拠です。
感覚器(眼・耳)への先回りの対応
生存例で長期的な生活の質を左右するのが、視覚と聴覚の進行性の障害です。COL11A1が原因のFBCG1では、眼と耳の合併症を避けることが難しいため、診断と同時に先回りの監視を始める必要があります。眼については、強度近視・白内障が幼少期から進むため定期的な視力・細隙灯検査が必須で、網膜剥離のリスクが非常に高いため散瞳下での精密な眼底検査をスケジュール化します。耳については、新生児聴覚スクリーニングを通過しても進行性の感音難聴のリスクがあるため、定期的な聴力検査で追跡し、必要に応じて補聴器や言語聴覚士による療育支援を早めに導入します。
7. 遺伝カウンセリングの意義
FBCG1の診断がついた後は、ご家族への丁寧な遺伝カウンセリングが重要です。私たちは特定の選択を勧めたり、結論を急がせたりすることはありません。中立的な立場で情報をお伝えし、決定はご家族に委ねます。主に扱う内容は以下のとおりです。
- ➤遺伝形式と再発リスク:多くは常染色体潜性遺伝で、両親が保因者の場合、次のお子さんが発症する確率は理論上25%です。一方、顕性遺伝や生殖細胞系列モザイクの例も報告されているため、家系ごとに丁寧な評価が必要です。
- ➤保因者への配慮:COL11A1の変異は、1本だけ持つ保因者にも軽い近視や難聴と関連する可能性が示されています。患者さん本人だけでなく、ご家族全体を視野に入れたフォローアップが望まれます。
- ➤出生前診断の選択肢:家系内ですでに変異が同定されている場合、絨毛検査・羊水検査 による確実な出生前診断が可能です。受ける・受けないも含めて、ご家族が納得して選べるよう支援します。
- ➤予後情報の共有:知能の発達には直接影響しないという事実は、将来の教育や生活設計を考えるうえで、ご家族にとって大きな希望の根拠になります。
8. よくある誤解
誤解①「必ず生まれてすぐ亡くなる」
かつてはそう考えられていましたが、積極的な呼吸管理によって学童期・思春期まで成長する例が報告されています。重症度には幅があります。
誤解②「知的障害を伴う」
FBCG1は知能の発達そのものには直接影響しないと考えられています。生存例では正常な知能が確認されています。
誤解③「親が健康なら遺伝ではない」
多くは両親が無症状の保因者で、親が健康でも子に発症する潜性遺伝です。「健康だから遺伝ではない」という思い込みが診断を遅らせることがあります。
誤解④「FBCG1とFBCG2は同じ」
骨格の見た目は似ていますが、原因遺伝子が異なります。1型(COL11A1)は近視・難聴を高頻度に伴い、2型(COL11A2)は一般に視覚障害を伴いません。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 希少疾患の診断・遺伝カウンセリングについて
線維軟骨発生症をはじめとする希少な遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] OMIM #228520. Fibrochondrogenesis 1; FBCG1. Johns Hopkins University. [OMIM]
- [2] Orphanet. Fibrochondrogenesis. ORPHA:2021. [Orphanet]
- [3] MedlinePlus Genetics. Fibrochondrogenesis. National Library of Medicine. [MedlinePlus]
- [4] Tompson SW, et al. Fibrochondrogenesis Results from Mutations in the COL11A1 Type XI Collagen Gene. Am J Hum Genet. 2010;87(5):708-712. [PMC2978944]
- [5] Tompson SW, et al. Dominant and Recessive Forms of Fibrochondrogenesis Resulting from Mutations at a Second Locus, COL11A2. Am J Med Genet A. 2012. [PMC3264686]
- [6] A Novel Dominant COL11A1 Mutation Resulting in a Severe Skeletal Dysplasia. PMC. 2022. [PMC9185704]
- [7] Annals of Pediatric Endocrinology & Metabolism. A long-term survivor of fibrochondrogenesis diagnosed by whole exome sequencing. Ann Pediatr Endocrinol Metab. [e-APEM]
- [8] Fibrochondrogenesis, an Antenatal and Postnatal Correlation. J Clin Imaging Sci. [PMC3307213]
- [9] GARD (Genetic and Rare Diseases Information Center). Fibrochondrogenesis. NIH/NCATS. [GARD]
- [10] NCBI MedGen. Fibrochondrogenesis 1 (Concept Id: C3278138). [NCBI MedGen]






