目次

第17番染色体異常のすべて:トリソミーから微小欠失・重複症候群まで徹底解説
ヒトの細胞には通常、両親から1本ずつ受け継いだ23対(計46本)の染色体が存在します。その中でも第17番染色体は、細胞内の全DNAのわずか約2.5%から3%を占める比較的小さな常染色体です。物理的な長さは約8,300万個のDNA塩基対(bp)に及びます。
しかし、この第17番染色体は物理的なサイズに対して遺伝子が非常に密集している「ジーンリッチな染色体」として知られています。タンパク質の合成をコードする遺伝子が約1,100から1,200個も存在し、初期胚発生、神経構築、代謝制御、および腫瘍抑制に関わる極めて重要な遺伝子群が含まれています。
🧬 第17番染色体に含まれる代表的な重要遺伝子
- TP53: 強力な腫瘍抑制遺伝子。がんの発生を防ぐ「ゲノムの守護神」。
- RAI1: 概日リズム(睡眠)や神経行動を制御する転写調節因子。
- PAFAH1B1 (LIS1): 胎児期の脳の神経細胞遊走(適切な場所への移動)に不可欠。
- HNF1B: 腎臓の発生や糖代謝(インスリン分泌など)に関与。
- KANSL1: クロマチン修飾に関わり、知的発達や言語形成に影響。
これほどまでに多様かつ重要な遺伝子群が密集しているため、第17番染色体における「数の異常(トリソミーやモノソミー)」や「構造の異常(微小欠失、微小重複、同腕染色体など)」は、細胞レベルでの深刻な遺伝子量(ジーン・ドーズ)の不均衡を引き起こします。この不均衡は、多発先天奇形、重篤な知的障害、特異な神経行動学的表現型、そして悪性腫瘍の発生と進行に直結するのです。
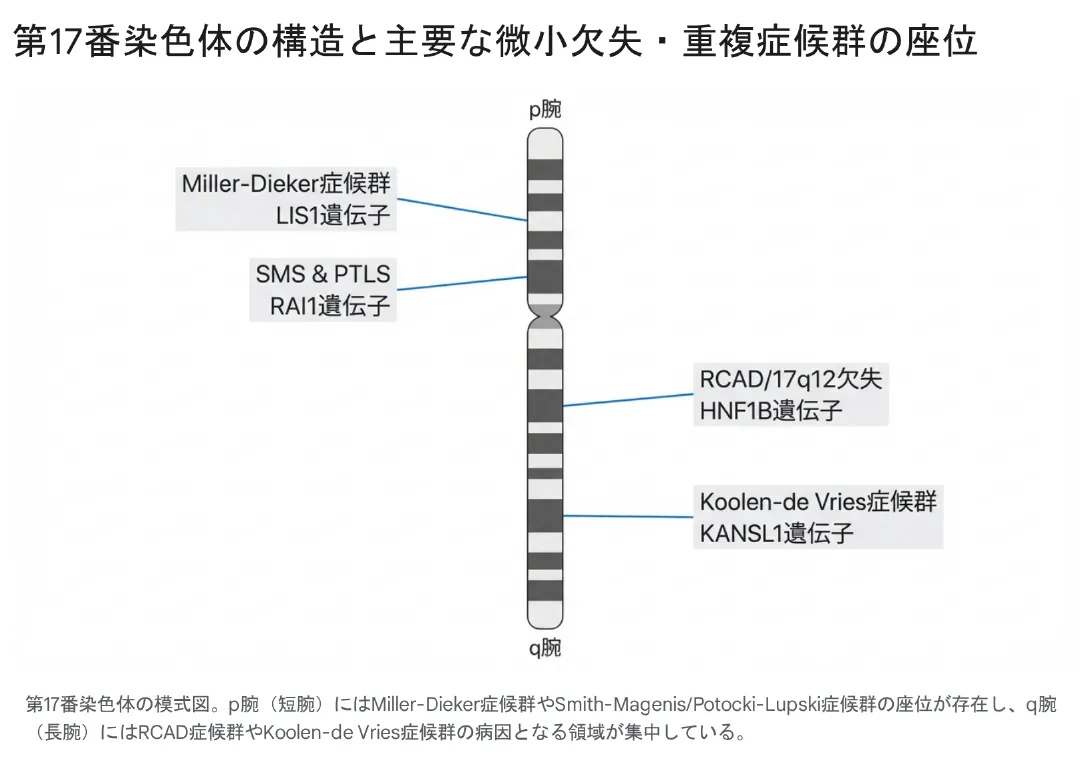
第17番染色体の構造模式図。p腕(短腕)とq腕(長腕)の各領域に様々な症候群の病因となる領域が集中しています。
1. 第17番染色体の全域的数的異常(トリソミー17)
細胞内に第17番染色体が3本存在する状態を「トリソミー17」と呼びます。この異常がすべての細胞に及ぶ「完全トリソミー」と、正常な細胞系列とトリソミーの細胞系列が混在する「モザイクトリソミー」とでは、臨床的帰結と生存可能性が根本的に異なります。
完全トリソミー17(Full Trisomy 17)の胎生致死性
完全な第17番染色体トリソミーは、細胞分裂時(多くは卵子や精子が作られる減数分裂期)における染色体不分離によって生じます。しかし、非常に重要な点として、生きた新生児として誕生した報告は医学文献上これまで一件も存在しません。
自然流産に至った胎児の約0.1%に検出されるのみであり、この事実は完全トリソミー17が着床後早期または胎生期において完全に致死的(embryonic lethal)であることを明確に示しています。これは、第17番染色体にコードされる多数の重要な遺伝子群の過剰発現が、初期の胚発生プログラムに対して非互換的であるためと推測されています。
モザイクトリソミー17(Mosaic Trisomy 17)の臨床像と予後
完全トリソミーが致死的であるのとは対照的に、正常細胞と異常細胞が混じる「モザイクトリソミー17」は、極めて稀ではあるものの、妊娠が継続し、出生に至り生存する症例が報告されています。過去の文献において羊水検査で出生前に発見された症例はわずか28例にとどまります。
その臨床的スペクトラムは「完全に正常な表現型」から「重篤な多発奇形を伴う致死的な状態」まで極めて多様です。この著しい多様性は、どの組織に、どの程度の割合でトリソミー細胞が分布しているか(組織特異的モザイク)に起因します。
羊水検査でモザイクが検出されても、胎児の超音波検査が完全に正常である場合があります。これは、トリソミー細胞が胎盤(羊膜など)にのみ存在し、胎児自身には存在しない限局性胎盤モザイク(CPM)の可能性が高いです。受精後のごく初期に、胎児へ発生する細胞から余分な染色体が排除される「トリソミーレスキュー」という自己修復機構が働いた結果と考えられています。
胎児期(出生前)の超音波検査で異常が観察される場合、以下のような所見が報告されています。
| 超音波所見のカテゴリー | 具体的な医学的特徴 |
|---|---|
| 発育と全身状態 | 子宮内胎児発育遅延(IUGR)、項部肥厚(Nuchal thickening)、嚢胞性ヒグローマ |
| 中枢神経系 | 小脳低形成、小脳囊胞 |
| 心血管系 | 心室中隔欠損症(VSD)などの心奇形、単一臍帯動脈 |
| 骨格・四肢・その他 | 非対称な奇形、足の体位異常、長管骨の短縮、胸水 |
出生後の真のモザイクトリソミー17患者は、非常に多岐にわたる合併症を呈します。神経・発達学的所見としては重度の知的障害や体幹の著明な筋緊張低下が見られます。生存した患者でも、14か月時点で6〜8か月の発達水準(喃語、手振り程度)にとどまるケースが報告されています。
身体的特徴としては、身体および顔面の左右非対称(Asymmetry)、広い前額部、眼内皆開離(離れた眼)、小顎症などが見られます。また、先天性気管気管支巨大症などの呼吸器異常による気管切開依存、冠動脈拡張などの心疾患、腎水腫、停留精巣など全身の器官に影響が及びます。
⚠️ 診断における重大な落とし穴:Gバンド法の偽陰性
生後診断において、末梢血リンパ球を用いた標準的な染色体検査(Gバンド分染法)を実施した場合、「正常な核型(偽陰性)」となることが非常に多いという事実があります。これは、細胞分裂が活発な血液系において、トリソミー細胞が自然に淘汰されてしまうためです。したがって、生後の確定診断の真のゴールドスタンダードは、皮膚生検による線維芽細胞(Fibroblast)の解析となります。
2. 第17番染色体の部分トリソミー(微小重複症候群)
第17番染色体の一部のみが過剰となる部分トリソミーは、多くの場合、減数分裂時における非アレル間相同組換え(NAHR: Non-allelic homologous recombination)という分子メカニズムによって引き起こされます。これは、類似したDNA配列間で誤った組み換えが起こり、一方の染色体には重複が、もう一方には欠失が生じるプロセスです。
17p11.2微小重複:ポトキ・ルプスキ症候群とは?17p11.2重複
Potocki-Lupski症候群(PTLS)は、第17番染色体短腕(17p11.2)の微小重複を原因とする先天異常症候群です。17p11.2重複症候群とも呼ばれます。1996年に初めて記述され、2007年にLorraine Potocki博士とJames R. Lupski博士によって包括的な臨床的記述が完了しました。有病率は全世界で約25,000人に1人と推定されています。
患者の約3分の2は、17p11.2領域における約370万塩基対(3.7Mb)の反復性のある重複を有しています。病態の核心は、この領域に含まれるRAI1遺伝子(Retinoic Acid-Induced 1)の量(ジーン・ドーズ)が過剰になることにあります。RAI1は他の遺伝子の活性を制御する転写調節因子として機能しています。
【PTLSの主要な臨床的特徴】
- 神経発達・行動的特徴: 乳児期の軽度から中等度の筋緊張低下(Hypotonia)、それに伴う嚥下障害が哺乳不良を引き起こします。認知障害(軽度から中等度の知的障害)が一般的です。行動面では、多動性、注意欠陥、不安が頻繁に見られ、多くの患者が強い自閉スペクトラム症(ASD)の基準を満たします。言語発達遅延や発話の失行も伴います。
- 身体・医学的特徴: 乳児期の成長障害(Failure to thrive)により、低体重・低身長の傾向があります。約40%の患者で先天性心疾患を合併します。また、睡眠時無呼吸症候群や入眠困難などの睡眠障害も高頻度で確認されています。
- 顔貌的特徴: 広い前額部と小顎症からなる三角形の顔立ち、下垂した眼裂(外側の目尻が下がる)、眼内皆開離などが特徴ですが、比較的軽微です。
17q12重複症候群
第17番染色体長腕の17q12領域における約1.4 MbのDNAが異常に重複する疾患です。この領域には少なくとも15の遺伝子が含まれていますが、特筆すべき点は「不完全浸透(Incomplete penetrance)」を示すことです。すなわち、この重複を持っていても明らかな症状が全く出ない人が存在します。症状が発現する場合は、知的障害、発達遅延、小頭症などが報告されています。
3. 第17番染色体の部分モノソミー(微小欠失症候群)
微小欠失症候群は、染色体の一部が失われることで、通常2コピー存在する遺伝子が1コピーとなる「ハプロ不全(Haploinsufficiency)」により発症します。第17番染色体には、医学的に極めて重要な微小欠失症候群が集中しています。
17p11.2微小欠失:17p11.2 Smith-Magenis症候群
Smith-Magenis症候群(SMS)は、PTLS(重複)と全く同じ17p11.2領域が「欠失」すること、またはRAI1遺伝子自体の病的変異によって引き起こされる複雑な神経行動学的疾患です。
🌙 特筆すべき臨床的特徴:睡眠の完全な逆転と特異行動
- 睡眠ホルモンの逆転: ほぼ100%の患者に見られます。睡眠を促す「メラトニン」の分泌リズムが完全に逆転し、日中にピークを迎え夜間に低下します。これにより、夜間の激しい中途覚醒と日中の強烈な眠気が引き起こされます。
- 自傷行為と特異な常同行動: 非常に愛情深い性格の一方で、爪や周囲の皮膚を執拗に剥がす「オニコチロマニア」、体腔(耳や鼻)に異物を挿入する「ポリエンボロコイラマニア」といった自己破壊的行動が見られます。また、指を舐めてページをめくる「lick and flip」や「self-hugging(自己抱擁)」は、SMSに極めて特異性の高い診断的指標です。
身体的・代謝的特徴: 幅広く四角い顔、テント状(M字型)の上唇、しゃがれ声が特徴です。また、興味深いことに17p11.2の「欠失」を持つ患者は脂質異常症(高コレステロール)になりやすく、RAI1の「変異」のみを持つ患者は高インスリン血症を伴う肥満になりやすいという明確な代謝的差異が報告されています。
📊 RAI1遺伝子量に依存する鏡像的表現型(Mirror traits)
同一ゲノム領域(17p11.2)における「欠失」と「重複」がもたらす、正反対の表現型の比較です。遺伝子量が人間の行動や発達にいかにダイナミックな影響を与えるかを示す、遺伝学上の重要なモデルとなっています。
| 比較項目 | Smith-Magenis症候群 (17p11.2欠失 / RAI1ハプロ不全) |
Potocki-Lupski症候群 (17p11.2重複 / RAI1過剰発現) |
|---|---|---|
| 睡眠パターン | メラトニンリズムの完全逆転、夜間覚醒、日中の強い眠気 | 睡眠時無呼吸症候群、入眠困難、睡眠維持の困難 |
| 行動特性 | 自傷行為、激しい癇癪、愛情深いが衝動的、注意を引く行動 | 多動、ASD(自閉スペクトラム症)傾向、引きこもり、不安 |
| 成長・身体 | 肥満傾向(腹部脂肪蓄積)、短頭、テント状の唇 | 成長障害、低体重・低身長、下垂した眼裂、三角形の顔 |
17p13.3微小欠失:ミラー・ディッカー症候群とは?17p13.3欠失と滑脳症
Miller-Dieker症候群(MDS:17p13.3微小欠失症候群)は、極めて重篤かつ致死的な遺伝性疾患です。病態の核心は、欠失領域に含まれるPAFAH1B1(一般にLIS1として知られる)遺伝子の喪失にあります。
LIS1遺伝子は、胎生期の脳発生において、神経細胞が脳の深部から表面へと適切に移動するプロセス(神経細胞遊走)に不可欠です。これが阻害されることで、大脳皮質の正常なシワが形成されず、脳の表面が異常に平滑になる「古典的滑脳症(Classical Lissencephaly)」を引き起こします。これにより、小頭症、重度〜最重度の知的障害、難治性のてんかん発作、深刻な嚥下障害が生じます。誤嚥性肺炎やてんかんのため、MDSの予後は非常に絶望的であり、幼少期を越えて生存できるケースはごくわずかです。
17q12微小欠失症候群:腎嚢胞糖尿病症候群(RCAD)
17q12欠失症候群は、主にHNF1B遺伝子とLHX1遺伝子の喪失により、多臓器にわたる症状を引き起こします。
- HNF1B遺伝子の喪失: 腎臓の構造的異常(嚢胞性異形成腎など)と、若年発症成人型糖尿病5型(MODY5:通常25歳以前に診断)が特徴的です。これらを合わせて「RCAD症候群」と呼びます。
- LHX1遺伝子の喪失: 脳の発達に影響を与え、統合失調症、ASD、ADHDなどの神経精神医学的疾患のリスクを増大させます。また、女性においては膣や子宮の発育不全を特徴とする「MRKH症候群」の直接的な原因となります。
17q21.31微小欠失:17q21.31微小欠失症候群(Koolen-de Vries症候群)
Koolen-de Vries症候群(KdVS)は、クロマチン修飾に関与するKANSL1遺伝子の機能喪失によって引き起こされます。
行動面で非常に特徴的なのは、大半の患者が極めて友好的で、愛想が良く、協調性がある(Friendly, amiable, cooperative)ことです。しかし、口腔の筋緊張低下と小児期の発語失行(CAS)が重なるため、言葉を発する(表出言語)ことに多大な困難を伴います。理解力(受容言語)は保たれているため、手話やAAC(補助代替コミュニケーション)機器の早期導入がフラストレーション軽減に極めて重要です。また、洋梨型の鼻(Pear-shaped nose)などの顔貌特徴やてんかんを伴いますが、成人期まで生存し社会生活を送ることが一般的です。
その他の重要な微小欠失
第17番染色体には、上記の他にも重要な疾患の病因領域が含まれます。例えば、神経線維腫症I型の重症化に関わるNF1欠失症候群とは?17q11.2微細欠失による「重症化しやすいNF1」や、末梢神経が圧迫に弱くなる17p11.2 遺伝性圧脆弱性ニューロパチー(HNPP)なども、この染色体上の異常に起因しています。
4. 構造異常と腫瘍学的意義:同腕染色体17q
第17番染色体は先天異常の要因となるだけでなく、後天的な細胞の体細胞変異を通じた「悪性腫瘍(がん)」の発生と進行においても極めて重要な役割を果たします。特に臨床的意義が高い構造異常が同腕染色体17q(Isochromosome 17q: i(17q))です。
細胞分裂の際、染色体は通常、縦方向(動原体)に綺麗に分離します。しかし、これが誤って「横方向」に分裂してしまうことがあります。第17番染色体でこれが起きると、短腕(p腕)が完全に失われ、長腕(q腕)が2つ複製されて融合した異常な構造を形成します。
細胞レベルで見ると、これは「17pの完全な喪失(モノソミー)」と「17qの部分的な過剰(トリソミー)」を同時に引き起こす複合異常です。17pの喪失は、強力な腫瘍抑制遺伝子であるTP53が失われることを意味し、同時に17qの重複は細胞増殖を促進する遺伝子群の増幅をもたらします。
この異常は、慢性骨髄性白血病(CML)やホジキンリンパ腫などの血液・造血器腫瘍において、病勢進行が早く治療抵抗性を示す予後不良因子となります。また、小児脳腫瘍である髄芽腫(Medulloblastoma)のGroup 4において頻繁に認められ、生存率を有意に低下させる強力なバイオマーカーとして機能しています。
5. 先進的診断技術と臨床的管理の進化
第17番染色体の構造的および数的異常を正確に特定するための診断技術は、過去数十年間で飛躍的な進化を遂げました。それぞれの検査法には明確な利点と限界が存在します。
- 非侵襲的出生前検査(NIPT): 母体血漿中のセルフリー胎児DNA(cffDNA)を利用し、トリソミー17や一部のコピー数増幅(CNV)をスクリーニングします。ただし確定診断には羊水検査が必要であり、CPMによる偽陽性リスクがあります。
- マイクロアレイ染色体検査(CMA/CNV-seq): ゲノム全体の微細な欠失や重複を高解像度で網羅的にスキャンする現代のゴールドスタンダードです。SMS、PTLS、MDSなどの診断に必須ですが、DNA量に変化がない平衡型転座は検出できません。
- FISH法: 特定の遺伝子座(MDSのLIS1やSMSのRAI1など)に対する蛍光プローブを用い、顕微鏡下で直接視覚化する技術です。事前に疑わしい疾患が絞り込まれている場合に極めて有効です。
- 核型分析(Gバンド分染法): 全体像を観察する古典的手法ですが、CMAに比べて解像度が低く(約5-10 Mb以下の異常は見逃す)、血液中のモザイク細胞は淘汰されるため偽陰性になりやすいという重大な弱点があります。
包括的医療と患者支援ネットワーク
第17番染色体に起因する疾患群は、いずれも単一の診療科にとどまらない多系統の複雑な異常を引き起こすため、小児科、臨床遺伝科、神経内科、循環器科などの包括的な多職種連携(Multidisciplinary approach)に基づく長期的な医学的管理が必須です。
運動機能向上を目指す理学療法(PT)、日常生活動作を支援する作業療法(OT)、そして言語聴覚療法(ST)の早期導入が不可欠です。また、国際的な「Chromosome Disorder Outreach (CDO)」や「Unique」、国内の「スミス・マギニス症候群患者会」など、希少疾患の家族が孤立を防ぎ、日々の療育ノウハウを共有するための社会的支援ネットワークへの接続が極めて重要です。
よくある質問(FAQ)
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
関連記事
参考文献
- Chromosome 17: MedlinePlus Genetics
- Potocki-Lupski syndrome – Genetics – MedlinePlus
- Smith-Magenis syndrome – Genetics – Medline Plus
- Miller-Dieker syndrome – Genetics – MedlinePlus
- 17q12 deletion syndrome – Genetics – MedlinePlus
- Koolen-de Vries Syndrome: What It Is, Symptoms & Treatment – Cleveland Clinic






