目次
- 1 1. β酸化とは:定義と歴史的背景
- 2 2. 脂肪酸の動員・細胞内取り込みと活性化
- 3 3. カルニチンシャトル:長鎖脂肪酸のミトコンドリア輸送システム
- 4 4. β酸化の4つの反応ステップ
- 5 5. エネルギー収支:パルミチン酸1分子から106 ATPが産生されるまで
- 6 6. 不飽和脂肪酸・奇数鎖脂肪酸の代謝:補助的な酵素機構
- 7 7. ペルオキシソームのβ酸化:ミトコンドリアとのクロストーク
- 8 8. β酸化の生理的役割:絶食・ケトン体・運動時代謝
- 9 9. β酸化異常症(FAODs)とMCADD:臨床的意義
- 10 10. 診断・遺伝子検査・治療の最前線
- 11 よくある質問(FAQ)
- 12 参考文献
- 13 関連記事

📍 クイックナビゲーション
脂肪酸β酸化は、私たちの体が脂肪をATPというエネルギー通貨に変換するための中心的な代謝経路です。空腹時・絶食中・長時間の運動時に最も重要性を増し、心臓・骨格筋・腎臓のエネルギーの大部分を担います。また、遺伝子の変異によってこの経路が障害されると、MCADDをはじめとする脂肪酸酸化異常症を引き起こし、新生児期から乳幼児期に生命を脅かす代謝クリーゼをもたらすこともあります。この記事では、β酸化の分子メカニズムから臨床疾患まで、基礎から臨床まで幅広く解説します。
Q. 脂肪酸β酸化とは何ですか?まず結論だけ知りたいです
A. 脂肪酸を2炭素ずつ切り出してアセチルCoAに変換し、最終的にATPを産生するミトコンドリアの異化経路です。1904年にKnoopが発見した古典的な経路で、パルミチン酸(炭素数16)1分子の完全酸化から106 ATPが得られます。MCADD等の遺伝的欠損による「脂肪酸酸化異常症(FAODs)」の基盤となる経路でもあります。
- ➤β酸化の定義と歴史 → 1904年Knoopの発見、β炭素酸化の命名由来
- ➤カルニチンシャトル → CPT1・CACT・CPT2による長鎖脂肪酸のミトコンドリア輸送機構
- ➤4つの反応ステップ → 脱水素化・水和・第二脱水素化・チオリシスの詳細
- ➤ATP産生量の計算 → パルミチン酸の完全酸化で106 ATPが産生される根拠
- ➤ペルオキシソームとのクロストーク → 両オルガネラの機能分担と代謝的連携
- ➤MCADD等の臨床疾患 → 病態・新生児スクリーニング・治療の最新知見
1. β酸化とは:定義と歴史的背景
脂肪酸β酸化(β-oxidation of fatty acids)は、細胞が脂肪酸からエネルギーを取り出すための主要な異化経路です。この反応はミトコンドリア・マトリックス内で行われ、脂肪酸の炭素鎖が2炭素ずつ切り出されてアセチルCoAが生成され、最終的にクエン酸回路(TCAサイクル)と電子伝達系を経て大量のATPが産生されます。
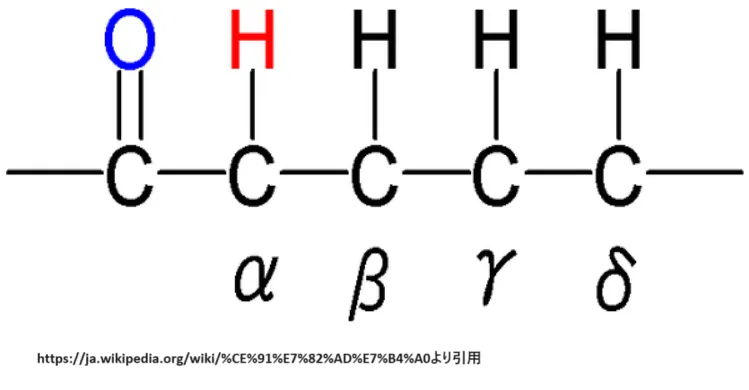
💡 用語解説:β炭素(ベータ炭素)
脂肪酸の炭素鎖において、カルボキシル基(-COOH)に最も近い炭素をα炭素(C2)、その隣の炭素をβ炭素(C3)と呼びます。β酸化という名称は、この「β炭素で酸化反応が起こる」という反応の本質を表しています。α炭素とβ炭素の間の結合が切断されることで、2炭素単位(アセチルCoA)が次々と切り出されていきます。
この代謝経路の歴史は1904年に遡ります。ドイツの医師・生化学者であるGeorg Franz Knoopが、標識された脂肪酸(フェニル基を付けた偶数鎖・奇数鎖の脂肪酸)を犬に投与した古典的な実験により、脂肪酸が2炭素単位で逐次的に分解されることを初めて明らかにしました。炭素数の偶数・奇数によって代謝産物が異なることからこの規則性が発見され、β炭素を標的とした酸化反応という概念が生まれました。現代生化学においても、この発見は脂質代謝研究の礎として評価されています。
絶食時・飢餓時・長時間の有酸素運動時など、グルコース(血糖)の供給が不十分な状況では、脂肪組織に蓄えられたトリアシルグリセロール(中性脂肪)が遊離脂肪酸として血液中に放出され、β酸化の基質として利用されます。骨格筋・心筋・腎臓のエネルギー需要の大部分がこの経路で賄われ、また肝臓でのβ酸化はケトン体産生(ケトジェネシス)を駆動して脳の代替エネルギー源を供給します。
2. 脂肪酸の動員・細胞内取り込みと活性化
β酸化が始まる前に、体は「エネルギーが不足している」というシグナルに応じて脂肪組織から脂肪酸を動員し、標的細胞へ輸送する精巧なシステムを稼働させます。
脂肪組織からの動員
絶食・運動・ストレス状態では、エピネフリン(アドレナリン)やグルカゴンといったホルモンが血中に増加します。これらのホルモンシグナルは脂肪細胞(脂肪組織)に存在する3つの主要な脂肪分解酵素——脂肪組織トリグリセリドリパーゼ(ATGL)・ホルモン感受性リパーゼ(HSL)・モノグリセリドリパーゼ(MGL)——を段階的に活性化します。これらの酵素の働きにより、脂肪組織に貯蔵されていたトリアシルグリセロール(TAG)が加水分解され、遊離脂肪酸(FFA)とグリセロールが血流へと放出されます。グリセロールは肝臓での糖新生の基質となり、遊離脂肪酸は血中アルブミンと結合して全身のエネルギー要求組織へと輸送されます。
細胞膜を通じた脂肪酸の取り込み
生理的pHでは脂肪酸は陰性荷電を持つため、単純拡散で効率よく細胞膜を通過することが難しく、複数の膜輸送タンパク質が協働して細胞内への取り込みを担います。代表的なものとして、脂肪酸輸送タンパク質(FATPs;FATP1〜6)・脂肪酸トランスロカーゼ(FAT/CD36)・細胞膜結合型脂肪酸結合タンパク質(FABPpm)があり、厳密な組織特異性を持っています。たとえばFATP1/FATP4は骨格筋に、FATP6は心筋に、FATP5は肝臓の脂肪酸取り込みに中心的な役割を担います。
アシルCoAへの活性化:代謝の入り口
細胞質内に取り込まれた遊離脂肪酸は、そのままでは代謝経路に入れません。アシルCoAシンテターゼ(ACS)という酵素がATPを消費して脂肪酸と補酵素A(CoA)を結合させ、高エネルギーのチオエステル結合をもつアシルCoAへと変換(活性化)します。この反応ではATPがAMPとピロリン酸(PPi)に分解され、続くピロリン酸の加水分解を含めると実質的にATP 2分子分のエネルギーが消費されます。この「初期投資」がのちのATP収支計算に関わります。
💡 用語解説:アシルCoA(Acyl-CoA)
補酵素A(CoA)はパントテン酸(ビタミンB5)を含む補酵素で、アシル基(脂肪酸由来の炭化水素鎖)と結合することで「アシルCoA」を形成します。このチオエステル結合は化学的に高エネルギーであり、後続の酵素反応を熱力学的に駆動する「活性化された」状態を意味します。β酸化・TCAサイクル・脂肪酸合成など、細胞内の脂質代謝の多くがアシルCoAを介して行われます。

3. カルニチンシャトル:長鎖脂肪酸のミトコンドリア輸送システム
短鎖・中鎖脂肪酸(炭素数12未満)は特別な輸送機構を必要とせずミトコンドリア内膜を直接通過できますが、生体内で最も豊富な長鎖脂肪酸(炭素数14〜20)のアシルCoA誘導体はミトコンドリア内膜を自由に通過できません。これを解決するのが「カルニチンシャトル(Carnitine Shuttle)」と呼ばれる精巧な輸送機構です。
💡 用語解説:カルニチンシャトル
L-カルニチンは肝臓・腎臓で生合成されるアミノ酸誘導体で、長鎖脂肪酸をミトコンドリア内膜の「内側」へ運ぶ「輸送体」として機能します。カルニチンが不足すると長鎖脂肪酸のβ酸化が障害され、心筋症・筋力低下・低血糖などの症状が現れることがあります(原発性カルニチン欠乏症)。食事では赤身肉・魚介類などに豊富に含まれています。
カルニチンシャトルの3つのコンポーネント
① CPT1
カルニチンパルミトイルトランスフェラーゼ1。ミトコンドリア外膜に局在。細胞質側のアシルCoAからアシル基をカルニチンに転移させ「アシルカルニチン」を生成。β酸化全体の律速段階(ボトルネック)として機能。摂食時はマロニルCoAによってアロステリックに阻害される。
② CACT
カルニチン-アシルカルニチントランスロカーゼ。ミトコンドリア内膜に埋め込まれたアンチポート(逆輸送体)。外側のアシルカルニチンをマトリックス内へ輸送し、同時に内部の遊離カルニチンを外へ排出する。SLC25A20遺伝子がコードする。
③ CPT2
カルニチンパルミトイルトランスフェラーゼ2。ミトコンドリア内膜のマトリックス側に位置。マトリックス内でアシルカルニチンをCoAと反応させ、再びアシルCoAと遊離カルニチンに分解。カルニチンはCACTで細胞質へ戻り再利用される。
⚠️ 重要な代謝調節:マロニルCoAによるCPT1阻害 摂食時、脂肪酸合成の中間体であるマロニルCoAが細胞内に蓄積すると、CPT1を強力に阻害します。これにより「脂肪酸の合成と分解が同時に起きる無益な回路(フタイルサイクル)」を防止します。絶食時にはマロニルCoAが低下し、この阻害が外れてβ酸化が活性化します。
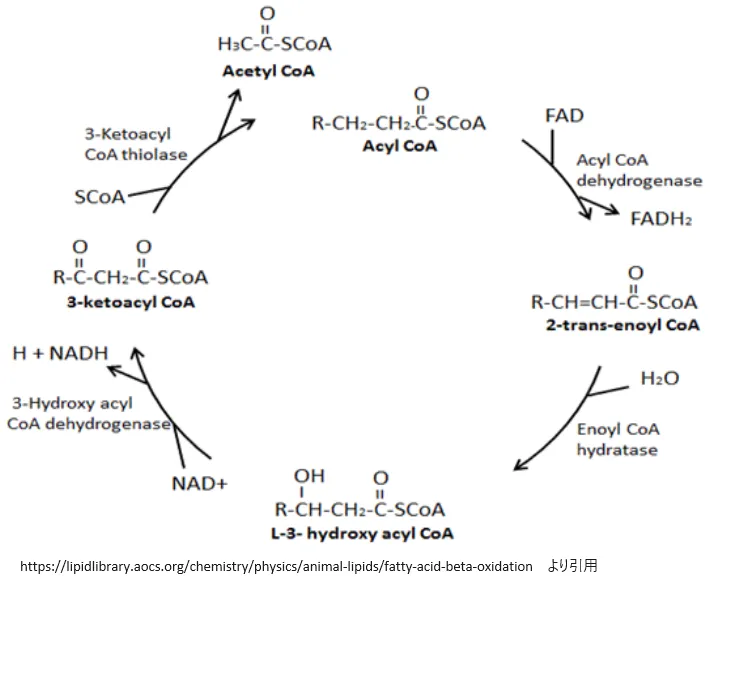
4. β酸化の4つの反応ステップ
ミトコンドリア・マトリックス内に取り込まれたアシルCoAは、4つの酵素反応からなるサイクル(スパイラル)に入ります。1サイクルごとにアシルCoAは炭素2つ分短くなり、1分子のアセチルCoA・1分子のFADH2・1分子のNADHが産生されます。このサイクルは、すべての炭素がアセチルCoAに変換されるまで繰り返されます。
| 反応ステップ | 触媒酵素 | 主要生成物 | 補酵素の変化 |
|---|---|---|---|
| ① 脱水素化 | アシルCoAデヒドロゲナーゼ | trans-Δ²-エノイルCoA | FAD → FADH₂ |
| ② 水和 | エノイルCoAヒドラターゼ | L-3-ヒドロキシアシルCoA | なし(H₂O付加) |
| ③ 第二脱水素化 | 3-ヒドロキシアシルCoAデヒドロゲナーゼ | 3-ケトアシルCoA | NAD⁺ → NADH + H⁺ |
| ④ チオリシス | β-ケトチオラーゼ | アセチルCoA + アシルCoA(n-2) | なし(CoA消費) |
各ステップの詳細解説
💡 ステップ①:脱水素化(アシルCoAデヒドロゲナーゼ)
アシルCoAデヒドロゲナーゼがα炭素(C2)とβ炭素(C3)の間から水素を2つ引き抜き、trans-Δ²-エノイルCoA(トランス型の二重結合を持つ構造)を生成します。この酸化に伴い、酵素の補欠分子族であるFADが還元されてFADH₂が形成されます。生成されたFADH₂は電子伝達フラボタンパク質(ETF)を介して直接ミトコンドリアの電子伝達系に電子を供与し、ATPの産生に貢献します。この酵素には脂肪酸の鎖長に応じた複数のアイソザイムがあります(次のインフォボックス参照)。
💡 用語解説:アシルCoAデヒドロゲナーゼのアイソフォーム(鎖長別)
脂肪酸の炭素鎖長によって異なる酵素が担当します。これらはアシル-CoAデヒドロゲナーゼファミリーとして分類されています:
ステップ②:水和 エノイルCoAヒドラターゼが第一ステップで生じた二重結合に水分子を付加し、β炭素にヒドロキシ基(-OH)を導入することでL-3-ヒドロキシアシルCoAを生成します。この反応はエネルギーの出入りなく(補酵素の変化なし)に進行し、次のステップへの足場を作ります。
ステップ③:第二脱水素化 3-ヒドロキシアシルCoAデヒドロゲナーゼが導入されたヒドロキシ基を酸化してケトン基(C=O)へと変換し、3-ケトアシルCoAを生成します。この酸化にNAD⁺が用いられ、NADH + H⁺が生成されます。NADHは電子伝達系で酸化され、2.5 ATP産生に貢献します。
ステップ④:チオリシス(加硫分解) β-ケトチオラーゼが新たな遊離CoAを用いてα炭素とβ炭素の間の結合を求核攻撃によって切断します。これにより脂肪酸鎖の末端から2炭素分がアセチルCoAとして遊離し、同時に元の基質より炭素数が2少ない短縮されたアシルCoAが生成されます。短縮アシルCoAはすべての炭素が分解されるまで再びステップ①から繰り返されます。
💡 用語解説:ミトコンドリア三頭酵素(MTP)
長鎖脂肪酸の酸化において、水和・第二脱水素化・チオリシスの3つの酵素活性は、ミトコンドリア内膜に結合したミトコンドリア三頭酵素(Mitochondrial Trifunctional Protein; MTP)と呼ばれる複合体によって効率的に担われます。MTPはHADHA遺伝子がコードするαサブユニット(LCEH・LCHAD活性)とHADHB遺伝子がコードするβサブユニット(LCKAT活性)からなるα₂β₂の四量体構造です。LCHAD欠損症(長鎖3-ヒドロキシアシルCoAデヒドロゲナーゼ欠損症)はMTPの障害から生じる疾患です。
5. エネルギー収支:パルミチン酸1分子から106 ATPが産生されるまで
β酸化がどれほどのATPを産生するかを理解するために、標準的なモデルとして炭素数16の飽和脂肪酸・パルミチン酸(C16:0)の完全酸化を計算します。
パルミチン酸(16炭素)を完全にアセチルCoAへ分解するには7サイクルのβ酸化が必要です。この7サイクルによって産生される還元型補酵素と代謝物は次の通りです:
- ➤アセチルCoA:8分子(7サイクル+最後の2炭素)
- ➤NADH:7分子(各サイクルのステップ③で生成)
- ➤FADH₂:7分子(各サイクルのステップ①で生成)
💡 用語解説:P/O比(プロトン-酸素比)
電子伝達系を通過する際に消費された酸素1原子あたりに産生されるATP分子数の比率です。現代の生化学的知見(プロトン勾配とATPシンターゼの化学量論に基づく厳密な計算)では、NADH 1分子→ 2.5 ATP、FADH₂ 1分子→ 1.5 ATPが学術標準値として採用されています。また、アセチルCoA 1分子がTCA回路を1周すると3 NADH・1 FADH₂・1 GTPが生成され、合計で10 ATP相当が得られます。かつての教科書ではNADH=3 ATP・FADH₂=2 ATPという整数値が使われていましたが、現在は非整数のP/O比に基づく計算が標準です。
パルミチン酸(C16)完全酸化における純ATP産生量の内訳
+80 ATP
+17.5 ATP
+10.5 ATP
= 108 ATP
− 2 ATP
106 ATP
※ 現代の学術標準値:NADH=2.5 ATP、FADH₂=1.5 ATP、アセチルCoA=10 ATP。旧来の教科書値(129 ATP)は古いP/O比に基づく計算。
グルコース(C6)の完全酸化で得られるATPが約30〜32分子であることと比べると、パルミチン酸(C16)から得られる106 ATPは圧倒的なエネルギー密度の高さを示しています。体重1kgあたりの貯蔵エネルギーとして、脂肪はグリコーゲン(糖質)の約6〜8倍の効率を持ちます。これが、長期の絶食や持久運動で脂肪が「最終的な燃料」として頼りにされる理由です。
6. 不飽和脂肪酸・奇数鎖脂肪酸の代謝:補助的な酵素機構
ミトコンドリアの主要なβ酸化酵素群は、飽和脂肪酸の代謝過程で形成されるトランス型の二重結合のみを基質として認識するよう進化的に最適化されています。しかし実際の食事に含まれる脂肪酸の多くは不飽和脂肪酸(シス型二重結合)であり、また一部の食物由来脂肪酸は奇数個の炭素を持ちます。これらを完全に分解するために補助的な酵素が動員されます。
不飽和脂肪酸の代謝
オレイン酸(C18:1)やリノール酸(C18:2)などの不飽和脂肪酸は、β酸化の途中でシス型の二重結合に遭遇すると酸化サイクルが一時停止します。これを克服するため2種の補助酵素が介入します。エノイルCoAイソメラーゼがシス型二重結合を酵素認識可能なトランス型へと異性化します。さらに多価不飽和脂肪酸(リノール酸・αリノレン酸など)では2,4-ジエノイルCoAレダクターゼがNADPHを消費して二重結合の還元・再配置を行い、β酸化が継続できる構造へと変換します。
奇数鎖脂肪酸の代謝:プロピオニルCoAの行方
奇数個の炭素を持つ脂肪酸(プロピオン酸・バレリン酸など、反芻動物の乳脂肪に多い)は、通常の4ステップのβ酸化スパイラルを繰り返しますが、最後のチオリシス反応で2炭素のアセチルCoAではなく3炭素のプロピオニルCoAが1分子残ります。
💡 用語解説:プロピオニルCoA → スクシニルCoAへの変換経路
プロピオニルCoAは以下の3ステップで代謝されます:
①プロピオニルCoAカルボキシラーゼ(ビオチン依存)→ D-メチルマロニルCoA
② メチルマロニルCoAラセマーゼ → L-メチルマロニルCoA
③ メチルマロニルCoAムターゼ(補酵素:ビタミンB12=コバラミン必須)→ スクシニルCoA
スクシニルCoAはTCAサイクルの直接の中間体であるため、奇数鎖脂肪酸は糖新生と脂質代謝を橋渡しする希有な経路を形成します。ビタミンB12欠乏ではこの変換が障害され、プロピオン酸/メチルマロン酸が蓄積します。
7. ペルオキシソームのβ酸化:ミトコンドリアとのクロストーク
哺乳類の細胞においてβ酸化はミトコンドリアだけで行われるわけではなく、単一膜の細胞小器官であるペルオキシソームでも並行して実行されています。この2つのオルガネラは似た4ステップ反応を共有しながらも、基質特異性・関与する酵素・生理的目的において明確に機能分担しています。
💡 用語解説:ペルオキシソーム
細胞質に存在する単一膜に囲まれた細胞小器官で、酵母・植物ではβ酸化の唯一の場所です。哺乳類では脂肪酸の極長鎖脂肪酸(VLCFA;炭素数22以上)・分岐鎖脂肪酸・プロスタグランジン・一部の胆汁酸中間体を優先的に処理します。ミトコンドリアのような複雑なカルニチンシャトルを必要とせず、VLCFAは直接膜を通過して内部で活性化されるのが特徴です。
| 機能的特徴 | ミトコンドリア | ペルオキシソーム |
|---|---|---|
| 主な基質 | 長鎖・中鎖・短鎖脂肪酸 | 極長鎖脂肪酸(C22以上)・分岐鎖 |
| 取り込み機構 | カルニチンシャトル(長鎖のみ) | カルニチン不要・直接取り込み |
| 第一段階の酵素 | アシルCoAデヒドロゲナーゼ | アシルCoAオキシダーゼ |
| 電子受容体 | FAD → 呼吸鎖 → ATP産生 | O₂ → H₂O₂(ATP非産生) |
| 生理的目的 | エネルギー産生(ATP) | VLCFA解毒・短縮・同化準備 |
⚠️ ペルオキシソームはATPを産生しない ペルオキシソームの第一段階はアシルCoAオキシダーゼが触媒し、引き抜いた電子を電子伝達系ではなく分子状酸素(O₂)に直接供与します。そのためATPは産生されず、代わりに活性酸素種の過酸化水素(H₂O₂)が生成されます。このH₂O₂はペルオキシソーム内に豊富に存在するカタラーゼによって直ちに水と酸素に分解・無毒化されます。
オルガネラ間の代謝的クロストーク
ペルオキシソームは長大な脂肪酸鎖をアセチルCoAまで完全に分解しきらず、炭素数が中鎖〜短鎖(C8前後)にまで短縮された段階で酵素反応が停止します。切り出されたアセチルCoAと短縮されたアシルCoA残基はカルニチンと結合してペルオキシソームから排出され、ミトコンドリアへと輸送されます。ミトコンドリアがこれらの中間代謝物を受け取り、TCA回路と呼吸鎖を用いて完全酸化とATP産生を完了させます。この巧みな分業と代謝的クロストークが、細胞全体の脂質毒性を防ぎ恒常性を維持する鍵です。
8. β酸化の生理的役割:絶食・ケトン体・運動時代謝
絶食・飢餓時:ケトジェネシス(ケトン体産生)の駆動
長期絶食・飢餓状態では、肝臓での糖新生(Gluconeogenesis)がTCA回路の中間体であるオキサロ酢酸を大量に消費します。同時に脂肪組織からの脂肪動員により肝臓のβ酸化が加速し、大量のアセチルCoAが産生されます。しかしオキサロ酢酸が枯渇しているためアセチルCoAはTCA回路に入れず、代謝的ボトルネックが発生します。
💡 用語解説:ケトン体(Ketone bodies)
肝臓が過剰なアセチルCoAから生成する水溶性の小分子。主な種類はアセト酢酸・D-β-ヒドロキシ酪酸・アセトンの3つです。遊離脂肪酸は血液脳関門(BBB)を通過できないため脳のエネルギー源になれませんが、水溶性のケトン体は血流で脳・心臓・骨格筋などへ輸送され、グルコース枯渇時の極めて重要な代替燃料として機能します。過酷な絶食状態では人体の総エネルギー消費量の最大20%をケトン体が担うとされています。未治療の1型糖尿病ではインスリン欠乏によりケトン体産生が制御不能となり、致死的な糖尿病性ケトアシドーシス(DKA)を引き起こします。
運動時:グルコース・脂肪酸サイクル(Randle Cycle)
長時間の有酸素運動では、骨格筋・心筋内の中性脂肪(IMTG)および血中から取り込まれた遊離脂肪酸由来のβ酸化が主要なエネルギー源となります。β酸化が活性化して大量のアセチルCoAやNADHが細胞内に蓄積すると、これらが解糖系の律速酵素やピルビン酸デヒドロゲナーゼ複合体をアロステリックに阻害します。この相互調節機構は1960年代にRandleらが提唱した「グルコース・脂肪酸サイクル(Randle Cycle)」として知られ、限られたグリコーゲン貯蔵量を節約する重要な生理的効果をもたらします。
9. β酸化異常症(FAODs)とMCADD:臨床的意義
β酸化経路を構成する酵素や輸送タンパク質の遺伝的欠損は、脂肪酸酸化異常症(FAODs:Fatty Acid Oxidation Disorders)と総称される先天性代謝異常症を引き起こします。これらは細胞内エネルギーの枯渇と毒性中間代謝物の蓄積という二重の病態をもたらします。
💡 用語解説:MCADD(中鎖アシルCoAデヒドロゲナーゼ欠損症)
MCADD(Medium-Chain Acyl-CoA Dehydrogenase Deficiency)は、FAODsの中で最も頻度が高い常染色体劣性遺伝性疾患です。北米・北西ヨーロッパ系集団では約1万5000人に1人、日本では約13万人に1人の頻度と推定されています。染色体1p31.1に位置するACADM遺伝子の変異(最多はc.985A>G、K304E変異)によって引き起こされ、中鎖脂肪酸(炭素数6〜12)を代謝するMCAD酵素が欠損します。
MCADDの病態カスケード:なぜ生命を脅かすのか
MCADD患者は通常の健康状態では完全に無症状ですが、乳幼児期の長時間絶食・発熱を伴うウイルス感染症・胃腸炎による嘔吐下痢などの生理的ストレス状態に直面すると、体がグリコーゲンを使い果たしβ酸化への依存度を急激に高めます。しかしMCAD酵素の欠損により炭素鎖が中鎖に短縮された段階で酸化サイクルが完全停止します。
⚡ 重篤な低血糖
脂肪酸からATPが得られないため肝臓の糖新生が停止し、急速に血糖値が低下。低血糖性脳症・けいれん・昏睡を引き起こす。
🔶 低ケトン性低血糖
アセチルCoAが生成されないためケトン体も作れず、脳の代替エネルギーが供給されない。「低ケトン性」は診断の重要な手がかり。
☠️ 毒性代謝物の蓄積
分解不能な中鎖脂肪酸(オクタン酸など)が肝臓・心臓・脳に異常蓄積し、ミトコンドリア機能不全・脳浮腫・急性肝障害を誘発。
⚠️ MCADD代謝クリーゼの症状:著しい嗜眠・筋力低下・反復性嘔吐を示し、適切な医療介入がなければ痙攣・昏睡・突然死(乳幼児突然死症候群:SIDSの一因)に至る。過去にはライ症候群(アスピリン関連急性脳症・肝障害)と誤診されるケースも少なくありませんでした。
その他の主要なFAODs
VLCAD欠損症
ACADVL遺伝子変異。極長鎖脂肪酸の酸化障害。MCADDの症状に加えて重篤な心筋症・反復性横紋筋融解症を呈することが多い。食事療法としてMCT(中鎖脂肪酸トリグリセリド)補充が必須。
LCHAD欠損症
HADHA遺伝子変異(MTPの一部)。長鎖3-ヒドロキシアシルCoA脱水素酵素の欠損。心筋症・末梢神経障害・網膜症を呈する。妊娠中に胎児がLCHADを持つ場合、母体にAFLP(妊娠中の急性脂肪肝)を引き起こすリスクがある。
SBCAD欠損症
ACADSB遺伝子変異。短鎖/分岐鎖アシルCoAデヒドロゲナーゼの欠損。イソバレリル基(2-メチルブチリル基)の代謝障害。詳細はSBCAD欠損症 疾患ページをご覧ください。
原発性カルニチン欠乏症
SLC22A5遺伝子変異。OCTN2カルニチントランスポーターの欠損。カルニチンが細胞内に保持できず長鎖脂肪酸のミトコンドリアへの移行が致命的に阻害される。心筋症・ミオパチーの原因となる。
10. 診断・遺伝子検査・治療の最前線
新生児マススクリーニングによる早期発見
💡 用語解説:新生児マススクリーニング(タンデム質量分析)
先進国の多くで導入されているスクリーニングプログラムです。生後数日以内に採取した乾燥血液濾紙(DBS)からタンデム質量分析(MS/MS)で特異的なアシルカルニチンプロファイルを測定し、発症前にFAODsを早期発見します。MCADDでは特にオクタノイルカルニチン(C8)の顕著な上昇が特徴的なバイオマーカーとなります。早期診断によって食事管理などの予防的介入が可能となり、代謝クリーゼや突然死のリスクを劇的に低下させることができます。
スクリーニング陽性の場合は、尿中有機酸分析(ジカルボン酸・ヘキサノイルグリシン・スベリルグリシン等の検出)・酵素活性測定(培養皮膚線維芽細胞や白血球を使用)・遺伝子解析(ACADM・ACADVL・ACADSB等)によって確定診断が行われます。
ミネルバクリニックで受けられる関連遺伝子検査
治療の基本原則
現在、MCADDに対する根治的な遺伝子治療は確立されていませんが、管理の基本は「長時間の絶食を絶対に避けること」というシンプルかつ効果的な原則です。
- ➤乳幼児期:頻回授乳(2〜3時間おき)・就寝前の炭水化物スナック摂取を徹底
- ➤急性疾患時:経口炭水化物摂取または点滴によるブドウ糖補充で異化亢進を速やかに逆転させることが救命の絶対条件
- ➤VLCAD・LCHAD欠損症:長鎖脂肪酸の厳格な制限と中鎖脂肪酸トリグリセリド(MCT)による代替エネルギー補充が必須
- ➤遺伝カウンセリング:常染色体劣性遺伝のため、両親は保因者(キャリア)。次子の出生前診断や家族全体の検査計画が重要。キャリアスクリーニングについて・米国人類遺伝学会の推奨もあわせてご覧ください
🏥 β酸化異常症・代謝疾患の遺伝子検査・遺伝カウンセリング
MCADD・VLCAD欠損症・LCHAD欠損症・SBCAD欠損症など
脂肪酸酸化異常症に関するご相談は、臨床遺伝専門医が在籍するミネルバクリニックへ
よくある質問(FAQ)
参考文献
- [1] Biochemistry, Fatty Acid Oxidation – StatPearls. NCBI Bookshelf. [NCBI]
- [2] Biochemistry, Lipolysis – StatPearls. NCBI Bookshelf. [NCBI]
- [3] Crosstalk between mitochondria and peroxisomes. PMC. [PMC4657118]
- [4] A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. PMC. [PMC2950079]
- [5] Carnitine transport and fatty acid oxidation. PMC. [PMC4967041]
- [6] Beta oxidation. Wikipedia. [Wikipedia]
- [7] Biochemistry, Ketogenesis – StatPearls. NCBI Bookshelf. [NCBI]
- [8] Medium-Chain Acyl-CoA Dehydrogenase Deficiency – StatPearls. [NCBI]
- [9] Medium-chain Acyl-CoA dehydrogenase deficiency: Pathogenesis, diagnosis, and treatment. PMC. [PMC9836253]
- [10] Role of Fatty Acids β-Oxidation in the Metabolic Interactions Between Organs. PMC. [PMC11641656]
- [11] Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. PMC. [PMC4729952]
- [12] A student centric method for calculation of fatty acid energetics. PubMed. [PubMed]
- [13] Biochemistry, Ketone Metabolism – StatPearls. [NCBI]
- [14] Fatty Acid beta-Oxidation. AOCS Lipid Library. [AOCS]






