目次

先天性腎性尿崩症2型(NDI2)は、AQP2遺伝子の変異により腎臓が尿を濃縮できなくなる希少疾患です。乳児期から1日数リットルにおよぶ大量の薄い尿が出続け、放置すると重度の高ナトリウム血症性脱水から不可逆的な知的障害を残すおそれがあります。一方で、早期診断と適切な水分・食事・薬物管理により、正常な成長と良好な腎機能を生涯維持できることもわかっています。この記事では、2024〜2025年に策定された国際専門家コンセンサスに基づく最新の診断・治療を、一般の方にもわかりやすく解説します。
Q. 先天性腎性尿崩症2型(NDI2)とはどのような病気ですか?
A. 腎臓の集合管にある水チャネル「アクアポリン2(AQP2)」の遺伝子変異により、抗利尿ホルモン(バソプレシン)の作用に応じて尿を濃縮できなくなる先天性疾患です。OMIM 125800、原因遺伝子はAQP2(12番染色体長腕12q13)。常染色体劣性型(AR-NDI2、約9%)と常染色体優性型(AD-NDI2、約1%)に分かれ、いずれも乳児期からの大量の薄い尿・反復する高ナトリウム血症性脱水・成長障害を呈します。
- ➤疾患の定義 → OMIM 125800、推定有病率は男性100万人あたり約8.8人
- ➤分子メカニズム → AR型は小胞体滞留、AD型はヘテロ四量体形成によるドミナントネガティブ効果
- ➤主な症状 → 著明な多尿(4mL/kg/時超)・高張性脱水・反復発熱・成長障害
- ➤診断 → 水制限試験+デスモプレシン負荷+AQP2/AVPR2遺伝子解析の統合
- ➤最新治療 → 国際コンセンサス2024に基づく水・食事・薬物療法と次世代創薬
1. 先天性腎性尿崩症2型(NDI2)とは:基本概念と疫学
先天性腎性尿崩症(Congenital Nephrogenic Diabetes Insipidus:NDI)は、腎臓が抗利尿ホルモン(バソプレシン)に応答できないために尿を濃縮することができず、大量の薄い尿が出続ける希少な遺伝性疾患です。同じ「尿崩症」という名前を持つ中枢性尿崩症(CDI)がホルモンの「分泌不全」であるのに対し、NDIはホルモンを「受け取って実行する」腎臓側の先天的な欠陥に起因します。
💡 用語解説:抗利尿ホルモン(バソプレシン/AVP)
脳の下垂体後葉から分泌されるホルモンで、体内の水分が不足したときに腎臓に「水を再吸収せよ」と指令を出す役割を担います。正式名称はアルギニン・バソプレシン(AVP)。AVPが腎臓の集合管にある受容体(AVPR2)に結合すると、細胞内で水チャネル「アクアポリン2(AQP2)」が細胞膜に移動し、尿から血液へ水を取り戻す仕組みが作動します。NDIではこの一連の流れのうち、受容体や水チャネル自体に問題があるため、ホルモンが正常に出ていても腎臓が応答できないのです。
NDIの3つのサブタイプ:NDI1とNDI2の違い
遺伝性NDIは、原因遺伝子と遺伝形式によって主に3つのサブタイプに分類されます。全体の約90%を占めるのがX染色体連鎖型のNDI1(原因遺伝子:AVPR2、OMIM 304800)であり、男性にほぼ限定して発症します。残りの約10%が本記事のテーマであるNDI2(原因遺伝子:AQP2、OMIM 125800)で、さらに常染色体劣性型(AR-NDI2、約9%)と常染色体優性型(AD-NDI2、約1%)に分かれます。
NDIの正確な有病率は世界的にも不明な点が多いものの、カナダ・ケベック州の疫学調査では男性100万人あたり約8.8人と推定されており、これが世界の一般的な有病率を反映していると考えられています。NDIという疾患概念は1947年にWilliamsとHenryによって医学文献上初めて提唱され、以後「バソプレシン抵抗性尿崩症」とも呼ばれながら、その分子病態の解明が飛躍的に進んできました。
2. 分子メカニズム:水を再吸収する精緻なシステム
NDI2の病態を理解するには、まず正常な尿濃縮の仕組みを知る必要があります。腎臓の集合管主細胞では、AVP-AVPR2-AQP2という3段階のリレーによって水の再吸収が精密に制御されています。
💡 用語解説:アクアポリン2(AQP2)
腎臓の集合管主細胞に特異的に発現する水の通り道(水チャネル)タンパク質です。細胞膜を6回貫通する独特の構造を持ち、4つの単量体が集まって四量体(ホモテトラマー)として機能します。普段は細胞内の貯蔵小胞にプールされていますが、AVPの刺激を受けると細胞膜の管腔側(尿が流れる側)へ一斉に移動し、尿から血液への水の再吸収を可能にします。
正常時のシグナル伝達:3段階リレー
血中のバソプレシン(AVP)が、集合管主細胞の基底膜側にあるV2受容体(AVPR2)に結合します。
Gタンパク質を介してアデニル酸シクラーゼが活性化され、細胞内のセカンドメッセンジャーcAMPが急増。これによりプロテインキナーゼA(PKA)が活性化されます。
PKAがAQP2のC末端のセリン256(Ser256)などをリン酸化すると、これがスイッチとなりAQP2四量体が貯蔵小胞から細胞膜(管腔側)へ移行。水の再吸収が一気に進みます。
💡 用語解説:cAMPとPKA
cAMP(環状アデノシン一リン酸)は細胞内で「ホルモンが届いた」というシグナルを伝える代表的な伝達物質(セカンドメッセンジャー)です。PKA(プロテインキナーゼA)はcAMPによって活性化される酵素で、標的タンパク質にリン酸を付ける(リン酸化する)ことで機能のスイッチを入れる役割を果たします。AQP2の場合、PKAによるリン酸化が細胞膜への移動の合図になります。
この一連のシステムには、Rab GTPaseファミリーやレトロマー複合体(Vps35)、SNAREタンパク質など、139種類にも及ぶ相互作用タンパク質が関与する精巧なネットワークが構築されています。AVPの刺激が24時間以上続く場合には、AQP2遺伝子の転写そのものが亢進し、細胞内のAQP2タンパク質の総量を増やす長期的な代償メカニズムも作動します。NDI2では、この精緻なシステムの最終実行役であるAQP2自体に異常が生じるのです。
3. AR型とAD型:同じ遺伝子なのに異なる病態
NDI2の最大の特徴は、同じAQP2遺伝子の変異でも、遺伝形式によって細胞内で起きる現象が全く異なることです。常染色体劣性型(AR-NDI2)と常染色体優性型(AD-NDI2)では、変異タンパク質の振る舞いも、臨床的重症度も大きく違います。
AR-NDI2(常染色体劣性型):小胞体での足止め
AQP2変異によるNDI2の約9割を占めるのがAR-NDI2です。患者は両親からそれぞれ変異したアレル(遺伝子のコピー)を受け継ぎ、ホモ接合体または複合ヘテロ接合体として発症します。両親はそれぞれ片方に変異を持つ「保因者(キャリア)」ですが、通常は無症状です。これまでに少なくとも52種類の病原性バリアントが報告されています。
💡 用語解説:小胞体(ER)滞留とミスフォールディング
細胞内でタンパク質が作られる場所を小胞体(Endoplasmic Reticulum:ER)といいます。タンパク質は正しい三次元立体構造に「折りたたまれて(フォールディングして)」初めて機能します。AR-NDI2では、変異したAQP2タンパク質がうまく折りたためず(ミスフォールディング)、細胞の品質管理システムによって異常品と判定され、小胞体から外に出してもらえず分解されてしまいます。これをER滞留と呼びます。本来あるべき細胞膜にAQP2が届かないため、水の再吸収ができなくなるのです。
変異の種類によって細胞内での運命はさまざまです。膜貫通領域のαヘリックス構造を破壊するL28PやS216P変異は構造を物理的に壊し、リン酸化部位そのものを失うS256L変異は活性化スイッチが入らなくなり、T126MやR187C、A147T変異は重篤な小胞体滞留を引き起こします。野生型AQP2タンパク質の半減期が約4時間であるのに対し、これらの変異型はわずか1.8〜2.7時間で急速に分解されてしまいます。
AD-NDI2(常染色体優性型):仲間を巻き込む「妨害」
AD-NDI2は全体の約1%という少数派です。片方のアレルにだけ変異があれば発症します。報告されている11種類以上の変異は、そのほとんどがAQP2タンパク質のC末端領域(行き先を指示するシグナルが含まれる領域)にあります。
💡 用語解説:ドミナントネガティブ効果
変異した異常タンパク質が、正常タンパク質の働きを「邪魔する」現象です。AQP2は4つの単量体が集まって四量体として機能するため、変異した単量体が1つでも混じった四量体(ヘテロ四量体)は、正しい行き先を見失ってしまいます。本来あるべき場所(管腔側細胞膜)ではなく、基底膜側に誤って運ばれたり、ゴルジ装置や分解経路に押し込まれたりするため、結果として水チャネルが正しく働けなくなります。
AD-NDI2のフレームシフト変異(763-772 delなど)では、C末端が異常に伸び、その伸びた部分に「基底膜側へ運ばれよ」というシグナル(チロシンベースのモチーフ)が新たに出現することが判明しています。意図せぬ目印が生まれるため、変異単量体を含む四量体は本来向かうべき管腔側ではなく逆方向の血管側へと誤輸送されてしまうのです。E258K変異では、ゴルジ装置やリソソームへの分解経路に流れてしまい、機能を発揮できません。
なぜAD型は軽症?──「16分の1の救世主」
AD-NDI2はAR-NDI2と比較して、発症が遅く(小児期後期や成人期初期の発症もある)、症状が軽度であることが知られています。これは確率論で説明できます。野生型と変異型の単量体が同量作られランダムに四量体を形成すると、16分の1(約6.25%)の確率で野生型のみで構成された純粋な正常四量体ができます。このわずかな確率で生まれる正常な四量体が、AVPに応答して正しく細胞膜へ移行するため、ある程度の残存水輸送能が維持されるのです。
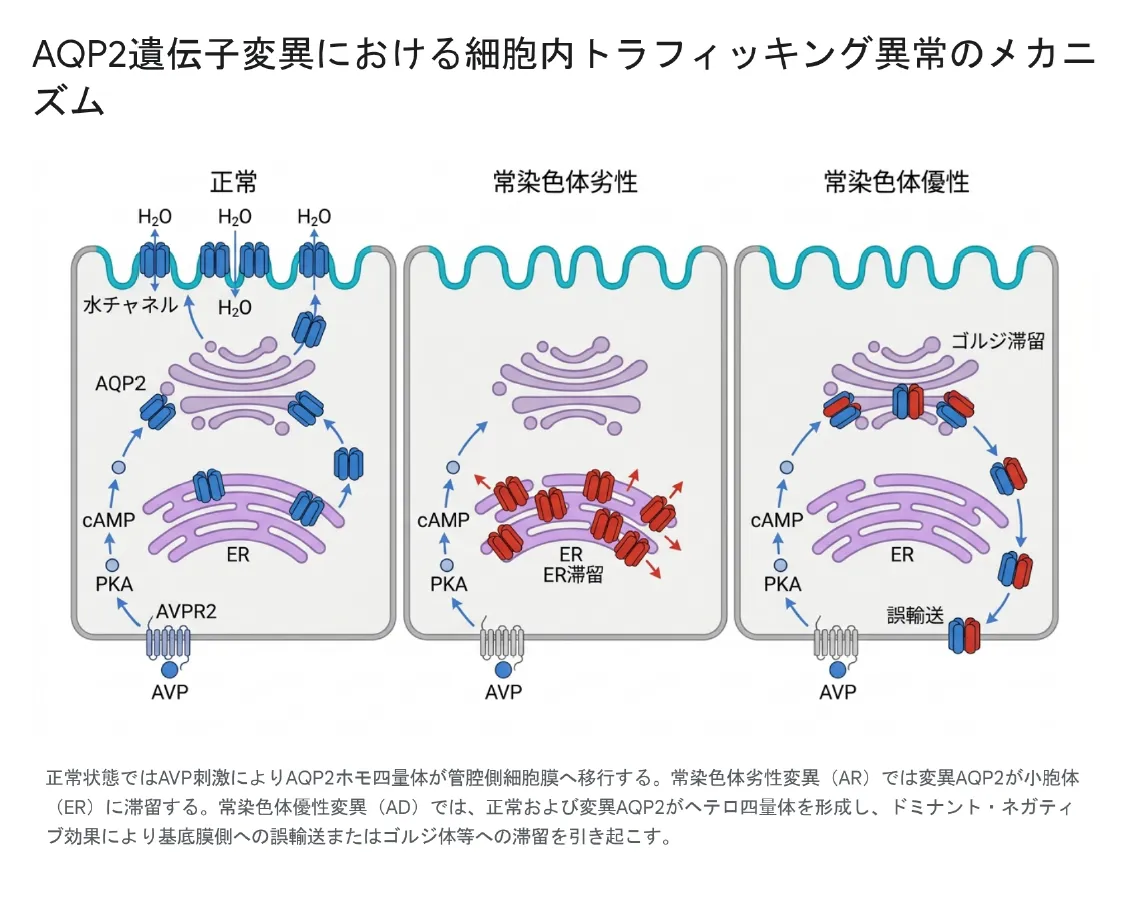
正常状態ではAVP刺激によりAQP2ホモ四量体が管腔側細胞膜へ移行する。常染色体劣性変異(AR)では変異AQP2が小胞体(ER)に滞留する。常染色体優性変異(AD)では、正常および変異AQP2がヘテロ四量体を形成し、ドミナントネガティブ効果により基底膜側への誤輸送またはゴルジ体等への滞留を引き起こす。

4. 主な症状:乳児期から始まる重篤なサイン
AR-NDI2の患者さんは、出生直後から極めて重篤な症状を呈します。乳児はまだ自分で「のどが渇いた」と訴えられないため、保護者や医療者が早期に異変を捉えることが重要です。
💧 著明な多尿と低張尿
成人で1日3リットル以上、小児でも2リットル以上、体重あたり4mL/kg/時を超える大量の極めて薄い尿を持続的に排泄します。尿比重は1.005未満と水に近い値です。
🚨 高ナトリウム血症性脱水
水分補給が尿喪失に追いつかず、急速に高張性脱水に陥ります。皮膚の乾燥、ツルゴール消失、眼球陥没、大泉門の陥凹、舟状腹(scaphoid abdomen)が見られます。
🌡️ 反復性発熱と消化器症状
脱水に伴う間欠的な高熱(脱水熱)が極めて一般的。水分を欲して過剰に飲むため、哺乳直後に嘔吐や空嘔吐(retching)を繰り返します。
📉 著しい成長障害
水分欲求のため胃が水で満たされ、十分なカロリー・栄養素を摂取できず、体重増加不良や低身長(Failure to Thrive)が顕著になります。
💡 用語解説:高ナトリウム血症(こうなとりうむけっしょう)
血液中のナトリウム濃度が異常に高くなった状態です。NDIでは大量の水だけが失われるため、体に残った血液は相対的に塩分濃度が高くなります。重症になると170 mmol/Lを超える危機的な数値に達することもあります。脳細胞が脱水・収縮し、けいれん発作・頭蓋内出血・昏睡を引き起こすため、緊急の補正が必要ですが、補正速度を間違えると逆に脳浮腫を起こす危険な病態です。
5. 診断と治療が遅れた場合の長期合併症
適切な治療介入が遅れると、生涯にわたる深刻な合併症を背負うリスクが高まります。逆にいえば、早期診断と適切な管理によってこれらの合併症の多くは予防可能です。
🧠 神経発達障害と知的障害
反復する重度の高ナトリウム血症性脱水は、脳細胞の収縮、頭蓋内出血、けいれん発作、昏睡を引き起こす可能性があります。脳への反復的な物理的・浸透圧的ダメージは、回復不能な知的障害・発達遅滞・言語遅延・自閉症スペクトラム障害・学習障害を残すリスクがあり、頭蓋内石灰化を伴うこともあります。
🫧 尿路系の物理的拡張
大量の尿の産生・排泄により尿路系に異常な高圧がかかり続け、巨大膀胱(Megacystis)・膀胱壁の肉柱形成・水尿管症(Hydroureter)・水腎症(Hydronephrosis)が高頻度に発生。反復性尿路感染症の温床となり、最終的には腎機能の物理的圧迫による低下を招きます。
🍽️ 慢性便秘と夜尿症
慢性脱水により大腸での水分過剰再吸収が起こり、小児期から頑固な便秘をもたらします。夜尿症(遺尿)も極めて一般的で、社会生活上の大きな負担となります。
📊 長期コホート研究の知見
英Great Ormond Street病院の36名コホート(追跡中央値9.5年)では、適切な治療により体重Zスコアが診断時-2.1から最終-0.2へ顕著に改善。一方で身長は-0.9と完全なキャッチアップが課題。eGFR(腎機能)は中央値81 mL/min/1.73m²と良好に維持され、原疾患が直接末期腎不全に進行するリスクは高くないことが示されています。
6. 診断:水制限試験から遺伝子検査まで
NDI2の確定診断は、臨床症状の評価・生化学的パラメータ・水制限試験・遺伝子解析を統合して確立されます。多尿・多飲・原因不明の発熱・成長障害・不適切な低張尿を呈する小児では、ただちにNDIを鑑別に挙げる必要があります。
尿崩症が「ほぼ確定」となる5つの所見
2024〜2025年の国際専門家コンセンサス・ステートメントは、以下の5つの生化学的所見が同時に確認された場合、尿崩症の診断はほぼ確定的(pathognomonic)であり、ただちに遺伝子検査を含む確定プロセスへ進むべきと明記しています。
- ①血清ナトリウム濃度:147〜150 mmol/L以上の正常高値〜高値
- ②血清浸透圧:300 mOsm/kgを超える高値
- ③尿浸透圧:高張に傾いているにもかかわらず200〜300 mOsm/kg未満の低張尿
- ④尿比重:1.005未満
- ⑤尿量:4 mL/kg/時を超える異常な多尿
水制限試験とデスモプレシン負荷
💡 用語解説:水制限試験とデスモプレシン
水制限試験は腎臓の尿濃縮能を動的に評価する検査で、医療従事者の厳格な監視下に水分摂取を制限し、血清・尿浸透圧の変化を経時的に測定します。続けてデスモプレシン(dDAVP:合成バソプレシンアナログ)を投与し、その後の尿浸透圧の変化を評価することで、中枢性(CDI)と腎性(NDI)を鑑別します。NDI患者では受容体や水チャネルの欠陥のためデスモプレシンを投与しても尿浸透圧の上昇は50%未満にとどまり、低張尿が持続します。
分子遺伝学的診断:AVPR2 → AQP2の順で解析
確定診断・予後予測・家族への遺伝カウンセリングのため、遺伝子パネル解析が不可欠です。臨床的なアルゴリズムでは、NDI症例の約90%を占めるAVPR2遺伝子をまずスクリーニング(シーケンシング、MLPA法など)し、病原性バリアントが同定されない場合に次のステップとしてAQP2遺伝子のシーケンシングへ進むのが標準です。男性発端者でAQP2のホモ接合体または複合ヘテロ接合体変異が同定されればAR-NDI2、ヘテロ接合体変異であればAD-NDI2と確定診断されます。
早期の遺伝学的診断は、不必要な侵襲的検査を回避し、先制的な水分・栄養管理を開始することで、不可逆的な知的障害や尿路合併症を未然に防ぐ最強のツールとなります。
7. 最新治療:2024年国際コンセンサスと次世代創薬
先天性NDIの根本的な遺伝子修復療法は現時点で実用化されていません。現在の治療目標は「尿量を安全かつ管理可能なレベルまで減らすこと」「水・電解質バランスを生涯維持すること」「正常な身体的成長と神経学的発達を保証すること」です。2024〜2025年に欧州希少腎疾患リファレンスネットワーク(ERKNet)と欧州小児腎臓病学会(ESPN)が策定した国際専門家コンセンサスが、現在の世界標準となっています。
水分管理:生涯にわたる「自由なアクセス」が最優先
急性期の脱水時には静脈内輸液が必要ですが、ここに重要な落とし穴があります。NDI患者の脱水は水分単独の喪失(フリーウォーター・ロス)が主体であるため、生理食塩水(0.9% NaCl)のような等張液の不用意な投与は、逆にナトリウム負荷を増やし高ナトリウム血症を悪化させる危険があります。推奨されるのは5%ブドウ糖液に0.225%塩化ナトリウムを加えた低張液による慎重な補給です。
食事療法:溶質負荷を減らす
食事療法の核心は、腎臓が排泄しなければならない「溶質負荷(オスモティック・ロード)」を減らすことです。臨床現場では【2×(Na+K, mmol)+ 4×タンパク質(g)】という簡便な計算式が用いられ、小児患者では1日あたり約15 mOsm/kg/日を目標に設定します。
💡 用語解説:溶質負荷(オスモティック・ロード)
食事から摂った塩分(ナトリウム・カリウム)とタンパク質を体が代謝した後、最終的に腎臓が尿に排泄しなければならない物質の総量のことです。腎臓は溶質を排泄するために必ず一定量の水を必要とするため、溶質負荷が多いほど尿量が増えてしまいます。NDIでは尿を濃縮できないため、この「水を引きずられる量」をいかに減らすかが治療の鍵になります。
ただし発育途上の小児ではタンパク質を極端に制限すると成長障害を悪化させるため、1日あたり2.0〜2.5 g/kgのタンパク質摂取は維持し、溶質負荷のコントロールは主に厳格な塩分制限で達成します。乳児期には母乳が標準フォーミュラより20〜30%、牛乳より約3.3倍も低負荷であるため、母乳栄養が最優先で推奨されます。重症例では夜間持続経管栄養(経鼻胃管・胃瘻)が必要となり、大規模コホートでは患者の約36%が乳児期から胃瘻造設などの介入を必要としたと報告されています。
薬物療法:「逆説的利尿薬」という妙手
国際的な専門医意識調査では、93%がチアジド系利尿薬、62%がアミロライド、55%がNSAIDsを処方の第一選択または併用薬として推奨しています。「尿崩症(多尿)の治療に利尿薬を使う」という一見矛盾するアプローチが、なぜ有効なのでしょうか。
💊 チアジド系利尿薬
遠位曲尿細管のNa-Cl共輸送体を阻害→軽度の塩分喪失と循環血液量減少→RAAS活性化→近位尿細管での水・ナトリウム再吸収が代償的に亢進。集合管に到達する原尿が減るため、結果として尿量が低下する精巧なメカニズム。低溶質食と併用すれば尿量を最大70%減少させられます。小児初期用量は1〜2 mg/kg/日。
💊 アミロライド(カリウム保持性)
チアジドの最大の副作用である低カリウム血症を相殺するため併用されます。集合管の上皮ナトリウムチャネル(ENaC)を阻害し、電解質バランスを安定させながら相乗的に尿量を減少。
💊 NSAIDs(インドメタシン等)
プロスタグランジン合成阻害により腎血流量とGFRを軽度低下、髄質浸透圧勾配を回復。ただし長期投与は不可逆的な間質性腎炎・消化管潰瘍のリスクがあり、最低限の用量・期間に留めるべきです。
長期コホートからの興味深い知見として、学童期以降に成長するとこれらの薬剤の有効性が外見上低下し、多くの患者で薬剤を完全離脱しても臨床的悪化が見られなくなる現象が報告されています。成長に伴う腎の代償機能の発達や、患者自身の水分摂取行動の自律的学習・適応が関与していると考えられています。
次世代創薬:分子病態に直接介入する標的治療
分子病態の解明が進んだことで、原因遺伝子やシグナル伝達経路の欠陥に直接働きかける標的治療の開発が世界中で精力的に進行しています。
🧪 化学的薬理シャペロン
グリセロール・DMSO・TMAOなどの低分子化合物が、ER滞留した変異AQP2の三次元構造を安定化し、品質管理をすり抜けさせる試み。T126MやR187C変異モデルで細胞膜への到達と機能回復が実証されています。
🧪 NDI-5033(AMPK活性化薬)
血糖値に影響を与えずにAMPKを選択的に活性化する新規低分子化合物。NephroDI Therapeutics社が開発中。前臨床で低血糖を起こさず用量依存的に尿濃縮能を改善、3週間連日投与でも薬剤耐性なし。第1/2相臨床試験への移行が準備中。
🧪 スタチン系(ドラッグリポジショニング)
脂質異常症治療薬として広く使われるスタチン類が、AVP非依存的にAQP2の管腔側局在を促進。シンバスタチンは健常者で尿浸透圧を有意に上昇させることが確認され、リチウム誘発性NDI患者ではアトルバスタチンの第2相試験も進行中。
🧪 PDE阻害薬(ロリプラム等)
cAMP分解酵素であるPDE4を阻害し、微弱なシグナルを増幅。AD-NDI2マウスモデル(763-772 del変異)で尿浸透圧を有意に上昇させ、優性変異への特異的治療として期待。PDE5阻害薬シルデナフィルの研究も進行中。
8. 遺伝カウンセリングと家族計画
NDI2の確定診断後、家族への丁寧な遺伝カウンセリングが必要です。AR型とAD型では家族の再発リスクが大きく異なります。
- ➤AR-NDI2の再発リスク:両親はそれぞれ保因者(無症状)であり、次子も同様に発症するリスクは25%、保因者となるリスクは50%。発症しない確率は25%です。
- ➤AD-NDI2の再発リスク:患者本人が子どもを持つ場合、変異が遺伝する確率は理論上50%。両親に同じ変異がない場合(de novo変異)には次子の再発リスクは低くなりますが、生殖細胞モザイクの可能性は完全には除外できません。
- ➤出生前診断・着床前診断の選択肢:家族内で病的バリアントが既に特定されている場合、次子妊娠時に羊水検査・絨毛検査による出生前遺伝子診断が選択肢として存在します。
- ➤結婚前のキャリアスクリーニング:常染色体劣性疾患では夫婦両方が同じ遺伝子の変異保因者であった場合に発症リスクが生じます。妊娠を考える前の段階で拡大保因者スクリーニングを受けることで、リスクを事前に把握できます。
- ➤長期管理体制と家族の心理サポート:生涯にわたる多専門科連携の管理が必要となるため、診断後も小児科・腎臓内科・内分泌科・臨床遺伝科などとの継続的な連携が重要です。
9. 臨床遺伝専門医からのメッセージ
NDI2は希少疾患でありながら、早期診断と適切な管理によって患者さんの将来が大きく変わる「介入価値の高い疾患」です。乳児期に「ミルクを飲んでもすぐに薄い尿が大量に出る」「原因不明の高熱を繰り返す」「体重が増えない」といった症状を見たとき、この疾患を鑑別の引き出しに入れておけるかどうかが、お子さんの一生を左右します。
特に、反復する高ナトリウム血症性脱水による不可逆的な知的障害は、診断と治療の遅れによって防ぎえなかった悲劇のひとつです。逆に、生後早期に診断がつき、水分への自由なアクセス・適切な食事療法・チアジド系利尿薬の併用が始まれば、多くの患者さんが正常な腎機能と良好な成長を維持できることが大規模コホート研究で示されています。「希少だから諦める」のではなく、「希少だからこそ専門医にたどり着く」ことが鍵となります。
ミネルバクリニックでは、AVPR2・AQP2を含む包括的な遺伝子検査と、結果説明から家族計画まで一貫した遺伝カウンセリングを提供しています。気になる症状やご家族の遺伝歴がある方は、ぜひお気軽にご相談ください。
よくある質問(FAQ)
🏥 希少疾患の診断・遺伝カウンセリングについて
先天性腎性尿崩症をはじめとする希少遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] OMIM #125800. Diabetes Insipidus, Nephrogenic, 2; NDI2. Johns Hopkins University. [OMIM]
- [2] Levtchenko E, et al. International expert consensus statement on the diagnosis and management of congenital nephrogenic diabetes insipidus (arginine vasopressin resistance). Nat Rev Nephrol. 2024. [PubMed 39438674]
- [3] Hereditary Nephrogenic Diabetes Insipidus. GeneReviews®. NCBI Bookshelf. [NCBI Bookshelf]
- [4] Hereditary Nephrogenic Diabetes Insipidus: Pathophysiology and Possible Treatment. Int J Mol Sci. 2017;18(11):2385. [MDPI]
- [5] AQP2: Mutations Associated with Congenital Nephrogenic Diabetes Insipidus and Regulation by Post-Translational Modifications and Protein-Protein Interactions. Cells. 2020;9(10):2172. [PMC7599609]
- [6] Long-term outcome in inherited nephrogenic diabetes insipidus. Clin Kidney J. 2019;12(2):180-187. [PMC6452213]
- [7] Current treatment of hereditary nephrogenic diabetes insipidus in children. Childhood Kidney Diseases. [chikd.org]
- [8] Pathogenesis and treatment of autosomal-dominant nephrogenic diabetes insipidus caused by an aquaporin 2 mutation. PNAS. 2006;103(17):6485-6490. [PNAS]
- [9] An AMPK activator as a therapeutic option for congenital nephrogenic diabetes insipidus. JCI Insight. 2021. [PMC8119225]
- [10] Effects of sildenafil, metformin, and simvastatin on ADH-independent urine concentration in healthy volunteers. Physiol Rep. 2018;6(7):e13665. [PMC5880873]
- [11] Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J Clin Invest. 2005. [PMC508814]
- [12] Orphanet. Nephrogenic diabetes insipidus, autosomal. [Orphanet]






