目次

📍 クイックナビゲーション
ネマリンミオパチー(NM)は、骨格筋の著しい筋力低下を主症状とする先天性ミオパチーの代表的な疾患です。原因となる遺伝子から、生涯にわたるケア、そして現在進行中の画期的な遺伝子治療の最前線まで、最新の医学的知見を網羅して解説します。
Q. ネマリンミオパチーとはどのような病気ですか?
A. 筋肉の収縮を制御する遺伝子に変異が起きることで発症する筋疾患です。
乳幼児期から呼吸不全を伴う重症型から、生涯を通じてゆっくり進行するタイプ、さらには成人期に急激に発症する特異なタイプまで、幅広い症状(臨床的異質性)を持つことが特徴です。
- ➤原因遺伝子 → NEBやACTA1など、12種類以上の遺伝子が関与
- ➤診断の要 → 網羅的な次世代シーケンサー(NGS)による確定診断が必須
- ➤成人発症型(SLONM) → 血液疾患としてのアプローチで劇的に回復する可能性
- ➤最新治療 → AAV遺伝子治療や既存薬(サルブタモール)の治験が進行中
1. ネマリンミオパチー(NM)とは?疾患の概要
ネマリンミオパチー(Nemaline Myopathy: NM)は、全身の骨格筋における筋力低下および筋緊張の低下(ハイポトニア)を主な症状とする、先天性ミオパチーの中で最も頻度の高い疾患群の一つです。
💡 用語解説:ネマリン小体(Rods / Nemaline bodies)とは?
本疾患の確定診断において最大の指標となる病理学的な特徴です。患者の筋組織を特殊な染色(ゴモトリクローム変法染色)で観察すると、細胞質内に「糸状」や「桿(かん)状」の異常なタンパク質の凝集体が見られます。この名称は、ギリシャ語で「糸」を意味する「nema」に由来しています。
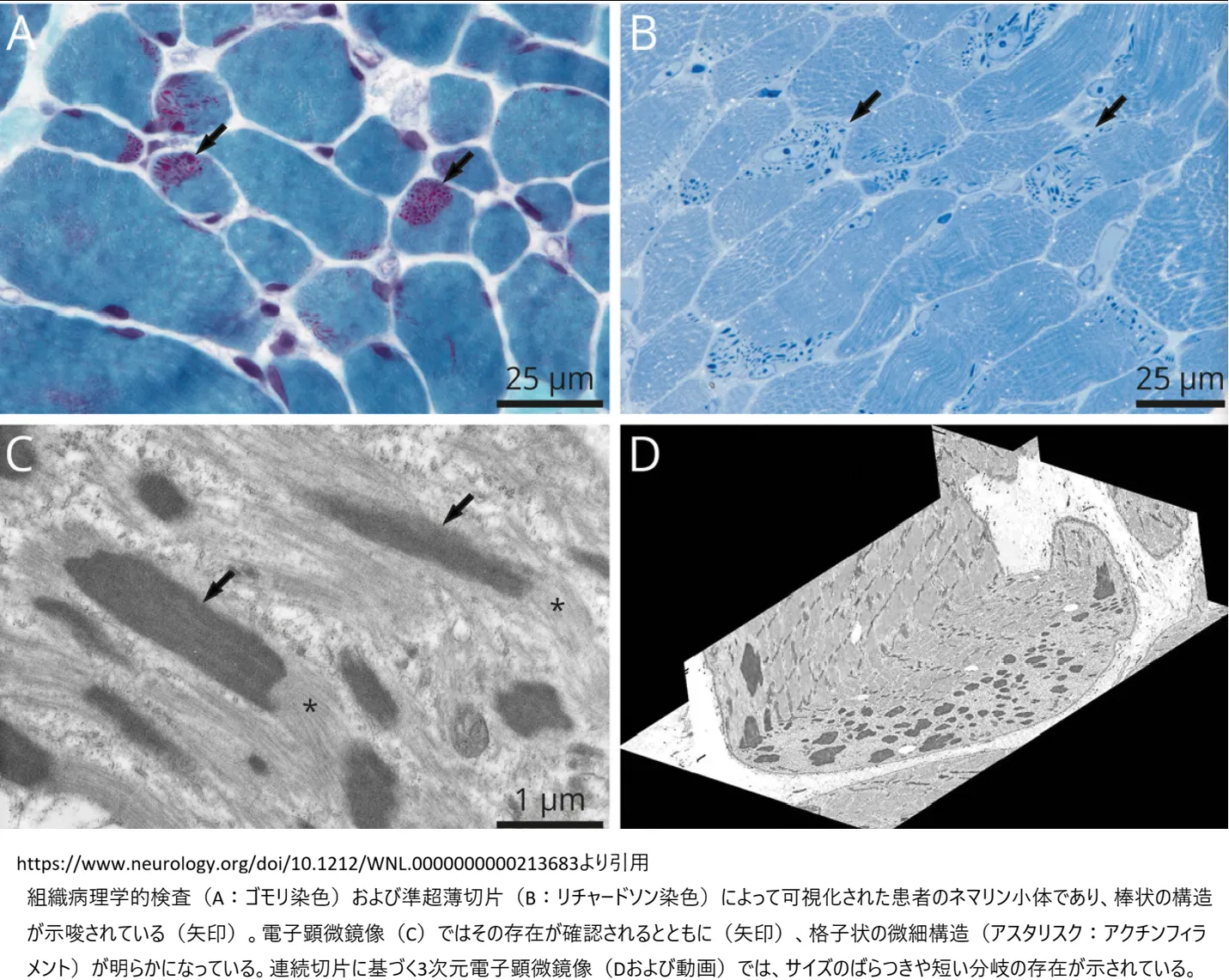
図:ネマリンロッドの形態像
組織病理学的検査(A:ゴモリ染色)および準超薄切片(B:リチャードソン染色)によって可視化された患者のネマリン小体であり、棒状の構造が示唆されている(矢印)。電子顕微鏡像(C)ではその存在が確認されるとともに(矢印)、格子状の微細構造(アスタリスク:アクチンフィラメント)が明らかになっている。連続切片に基づく3次元電子顕微鏡像(Dおよび動画)では、サイズのばらつきや短い分岐の存在が示されている。
(出典:Neurology, DOI: 10.1212/WNL.0000000000213683 より引用)
2. なぜ起こるのか?原因遺伝子とメカニズム
ネマリンミオパチーは、これまでに少なくとも12種類以上の異なる遺伝子に変異が生じることが原因で発症することが特定されています。これらの遺伝子はすべて、筋肉を収縮させるための基盤となるタンパク質の構築や制御に関わっています。
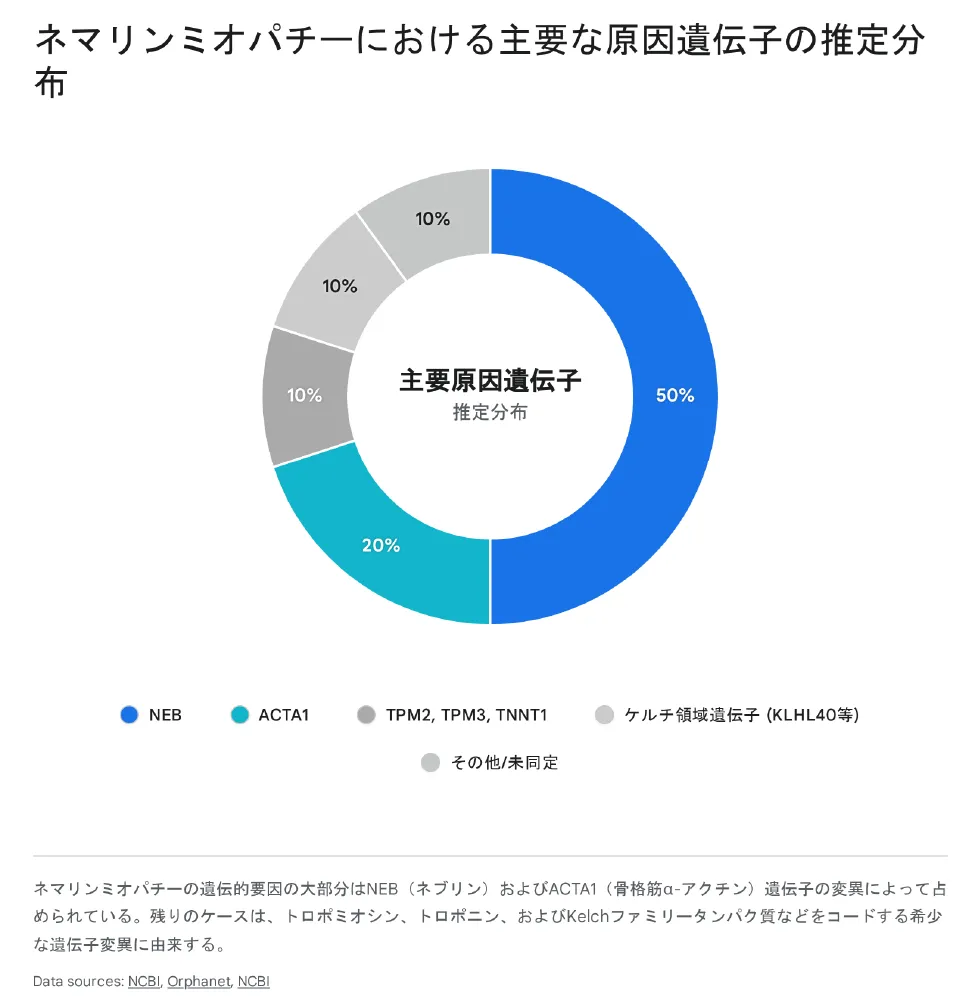
図1:ネマリンミオパチーにおける主要な原因遺伝子の推定分布
ネマリンミオパチー(Nemaline Myopathy: NM)の発症に関与することが知られている代表的な12個の原因遺伝子は以下の通りです。
🧬 ネマリンミオパチーの主な原因遺伝子(12個)
📌 補足情報
- 発症頻度の高い遺伝子: 上記の中でも、NEB(全体の約50%)と ACTA1(約20%)の変異が大部分を占めています。
- 関連タンパク質の役割: これらの遺伝子の多くは、骨格筋の収縮単位である「サルコメア(筋節)」を構成する「細いフィラメント(Thin filament)」の形成や機能、安定化に関わるタンパク質をコードしています。ここに異常が生じることで、筋収縮が正常に行われなくなります。
💡 用語解説:サルコメア(筋節)と細いフィラメント
サルコメアは、骨格筋の基本となるミクロの収縮単位です。ネマリンミオパチーの原因遺伝子は、このサルコメア内に存在する「細いフィラメント(Thin filament)」と呼ばれる構造の安定性や、筋肉を動かす際のカルシウム制御に不可欠な役割を担っています。ここが機能不全に陥ることで、効率的な筋収縮ができなくなります。
主要な原因遺伝子:NEBとACTA1
上の図1が示すように、最も高頻度で見られる原因遺伝子は以下の2つであり、これらで全体の約7割を占めます。
- ➤NEB遺伝子(ネブリン):全体の約50%
ネブリンは骨格筋に特異的な超巨大タンパク質で、アクチンフィラメントの長さを正確に決める「分子の定規」として機能します。常染色体劣性遺伝のパターンを示します。 - ➤ACTA1遺伝子(骨格筋α-アクチン):約15〜20%
細いフィラメントの主要構成要素そのものです。重篤な新生児期発症型においては、両親からの遺伝ではなく、受精卵の発育過程で偶然生じる「新生突然変異(De novo mutations)」として見つかることが多くあります。
新たなパラダイム:Kelch(ケルチ)ファミリー
近年、新たな病態メカニズムとして「Kelchファミリータンパク質(KLHL40、KLHL41など)」の関与が注目されています。これらは筋肉の構造そのものではなく、筋肉のタンパク質を異常な分解から保護する「シャペロン様」の役割を担っています。この保護機能が失われると、筋肉の構造が二次的に崩壊し、胎児期からの無動や重篤な症状を引き起こします。
📊 遺伝形式(AD/AR)による原因遺伝子の分類
ネマリンミオパチーは、同じ遺伝子でも変異のタイプによって遺伝形式が異なるケースがあるのが臨床上の大きな特徴です。実臨床では主に以下の3つのグループに分類して考えます。
1. 主に【AR】(常染色体潜性遺伝)を示す遺伝子(7個)
ご両親が保因者(キャリア)であり、変異アレルを2つ受け継いだ場合に発症します。
KLHL40
KLHL41
LMOD3
TNNT1
CFL2
MYPN
2. 主に【AD】(常染色体顕性遺伝)を示す遺伝子(1個)
変異アレルを1つ持つだけで発症します。KBTBD13は進行が遅く特異なタイプ(NEM6)の原因となります。
3. 【AD】と【AR】の両方が報告されている遺伝子(4個)
臨床現場で最も注意が必要なグループです。特にACTA1は、ご両親からの遺伝ではなく、受精卵での突然変異(デノボ変異)としてお子さんに発症するADパターンが大部分(約90%)を占めます。
TPM2
TPM3
TNNT3
ネマリンミオパチーの代表的な原因遺伝子は上記の12個ですが、臨床現場では「この12個を調べただけでは原因が特定できないケース」にしばしば遭遇します。
1. 新たな原因遺伝子の発見
近年の研究により、MYO18B(ミオシン18B)や RYR3(リアノジン受容体3)など、非常に稀な新しい原因遺伝子が次々と報告されています。
2. 他のミオパチーとの「オーバーラップ(重複)」
筋生検(病理検査)で「ネマリン小体」が見つかったとしても、それが必ずしも上記12個の遺伝子変異によるものとは限りません。他の先天性ミオパチーの原因遺伝子に変異がある場合でも、筋肉にネマリン小体が現れる(二次的な変化)ことが知られています。
- RYR1:本来は「セントラルコア病」の代表的な原因遺伝子ですが、コアとネマリン小体の両方が見られることがあります。
- SELENON(旧 SEPN1):本来は「マルチミニコア病」などの原因遺伝子です。
- MTM1 / DNM2:本来は「ミオチュブラーミオパチー / 中心核病」の原因遺伝子です。
だからこそ、確定診断には特定の少数の遺伝子だけをターゲットにするのではなく、次世代シーケンサー(NGS)を用いた「網羅的な遺伝子パネル検査」が不可欠なのです。
3. ネマリンミオパチーの6つの臨床サブタイプ
本疾患は臨床的なスペクトラム(多様性)が極めて広く、発症年齢や重症度に基づいて主に6つのサブタイプに分類されます。以下の図2は、発症年齢と疾患の重症度の相関を視覚的に表したものです。この分類は、長期的な予後の推定やケアの計画に不可欠です。
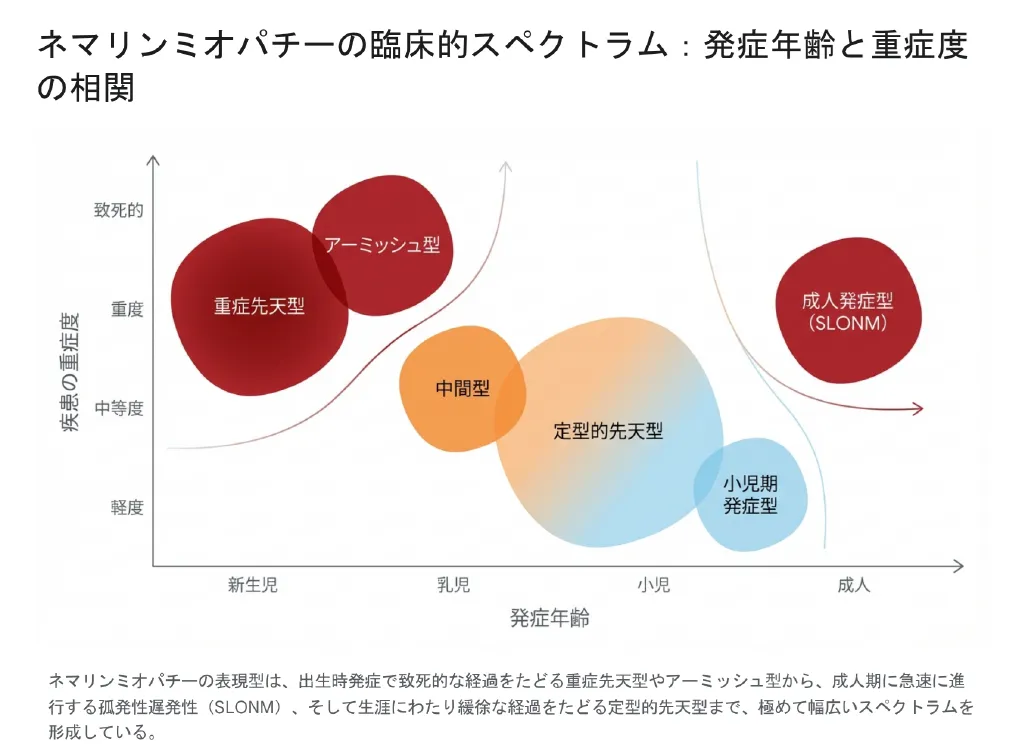
図2:ネマリンミオパチーの臨床的スペクトラム(発症年齢と重症度の相関)
1. 重症先天型(Severe Congenital)
図の左上(新生児期・致死的/重度)に位置し、全体の10〜20%を占めます。出生直後から重度の筋緊張低下(フロッピーインファント)や自発運動の欠如が見られ、嚥下障害と重篤な呼吸不全を伴います。人工呼吸器管理が必須となるケースが多くなります。
2. 定型的先天型(Typical Congenital)
図の中央(乳児期・中等度〜軽度)に位置する、最も一般的(約50%)な形態です。顔面や近位筋の筋力低下が見られますが、病状は時間経過とともに比較的安定しており、多くの場合、独立歩行を獲得します。寿命は健常者に近いことが多いとされています。
3. 中間型(Intermediate Congenital)
重症型と定型的の中間に位置します。運動発達が非常に遅く、多くの場合11歳頃までに自立歩行が困難となり、車椅子の使用や持続的な呼吸補助が必要になります。
4. 小児期発症型(Childhood-onset)
図の右下(小児期・軽度)に位置します。幼少期はほぼ正常に発達しますが、10歳前後に足首の背屈障害(下垂足)や対称性の筋力低下として発症します。進行は非常に緩徐です。
5. 成人発症型(Adult-onset / SLONM:Sporadic Late-Onset Nemaline Myopathy)
図の右上(成人期・重度〜致死的)に位置する特殊なタイプです。通常は家族歴のない孤発性として20代〜50代で発症します。急速に進行する体幹の筋力低下や重篤な呼吸不全を特徴とし、他のサブタイプとは全く異なる病態を示します(詳細は後述)。
6. アーミッシュ型(Amish Type)
特定の集団におけるTNNT1遺伝子変異に起因する、極めて稀で致命的なタイプです。
患者さんとご家族が一番知りたいこと:生命予後とこれからの生活
「この病気と診断されたら、寿命はどうなるのだろう?」「普通に学校に行ったり、生活したりできるのだろうか?」——これは、診断を受けた方やご家族が最も強く抱く、当然の不安です。
ネマリンミオパチーの生命予後は「どのサブタイプに該当するか」によって全く異なります。一概に「この病気だからこうなる」と言い切れないのが特徴です。
📊 サブタイプ別の一般的な生命予後
-
① 重症先天型の場合
自発呼吸や嚥下(飲み込み)が極めて難しいため、残念ながら生後1年以内での致死率が高いのが現状です。しかし、早期からの人工呼吸器管理と胃瘻(いろう)などによる栄養サポートにより、生命予後を延ばす取り組みが行われています。
-
② 定型的先天型(最も多いタイプ)の場合
乳幼児期の呼吸器感染症などを乗り越えれば、多くの場合、生命予後は健常な方と大きく変わりません。筋力の低下はゆっくりとしたものであり、歩行を獲得し、学校生活を送り、社会人として活躍されている患者さんも数多くいらっしゃいます。
-
③ 成人発症型(SLONM)の場合
無治療のままだと呼吸不全により1〜5年で致命的になる恐れがありますが、前述の通り、血液疾患としての適切な治療(自己幹細胞移植など)を早期に行うことで、劇的な回復と長期生存が見込めるようになっています。
日常生活の質(QOL)を守るために
定型的先天型や軽症型の場合、多くの子どもたちが地域の学校に通い、学業や趣味を楽しんでいます。ただし、日常生活において以下の点に配慮することが、長期間QOLを維持する鍵となります。
過度な疲労を伴う激しい運動は避けるべきですが、筋力を維持するための適度なストレッチや水泳などは推奨されます。学校の体育では「見学」だけでなく、できる範囲での参加方法を学校側と相談することが大切です。
筋力が弱いことで「咳をする力」が弱く、ただの風邪から重症の肺炎に進行するリスクがあります。インフルエンザやRSウイルスなどの予防接種を徹底し、体調不良時は早めに受診する体制を整えましょう。
4. 診断と次世代シーケンサー(NGS)による遺伝子検査の重要性
先天性ミオパチーの鑑別診断には、多角的なアプローチが必要です。初期評価では、クレアチンキナーゼ(CK)値の測定(ネマリンミオパチーでは正常〜軽度上昇)や、筋肉の超音波・MRI画像による特異的なパターンの確認が行われます。
確定診断のプロセス
従来から、診断のゴールドスタンダードは「筋生検」であり、ゴモトリクローム染色によってネマリン小体を同定することが決定打となります。しかし、現在の診断プロトコルにおいて最終的な確定を行うためには、次世代シーケンサー(NGS)を用いた網羅的遺伝子パネル検査や全エクソーム解析が不可欠です。
⚠️ なぜNGSによる遺伝子診断が重要なのか?
原因遺伝子(NEB、ACTA1など)を正確に特定することは、今後の進行予測や遺伝カウンセリングに直結します。さらに重要なのは、将来的に特定の遺伝子をターゲットとした最新の遺伝子治療の治験へ参加するための「必須条件」となるためです。
ミネルバクリニックのネマリンミオパチー遺伝子検査
当院では、次世代シーケンサー(NGS)を用いた網羅的な「ネマリンミオパチー遺伝子検査パネル」を提供しています。原因遺伝子の特定から確定診断、将来の治療選択肢の検討まで、臨床遺伝専門医がしっかりサポートいたします。
5. 孤発性遅発性ネマリンミオパチー(SLONM):治療パラダイムの転換
小児期の遺伝性タイプとは全く異なる概念として対応すべきなのが、成人期以降に急速に発症する孤発性遅発性ネマリンミオパチー(SLONM)です。
SLONMを発症した患者の半数以上が、MGUS(意義不明のモノクローナルガンモパチー)という血液異常を伴っていることが明らかになっています。MGUSを伴うSLONMは非常にアグレッシブに進行し、首下がり(Head drop)や呼吸器麻痺へと急速に悪化します。
血液学的な治療アプローチによる劇的な回復
近年、この疾患を「自己免疫疾患」ではなく「悪性腫瘍に準ずる形質細胞疾患」として捉え直すパラダイムシフトが起きました。以下の図3は、その最新の治療アルゴリズムを示しています。
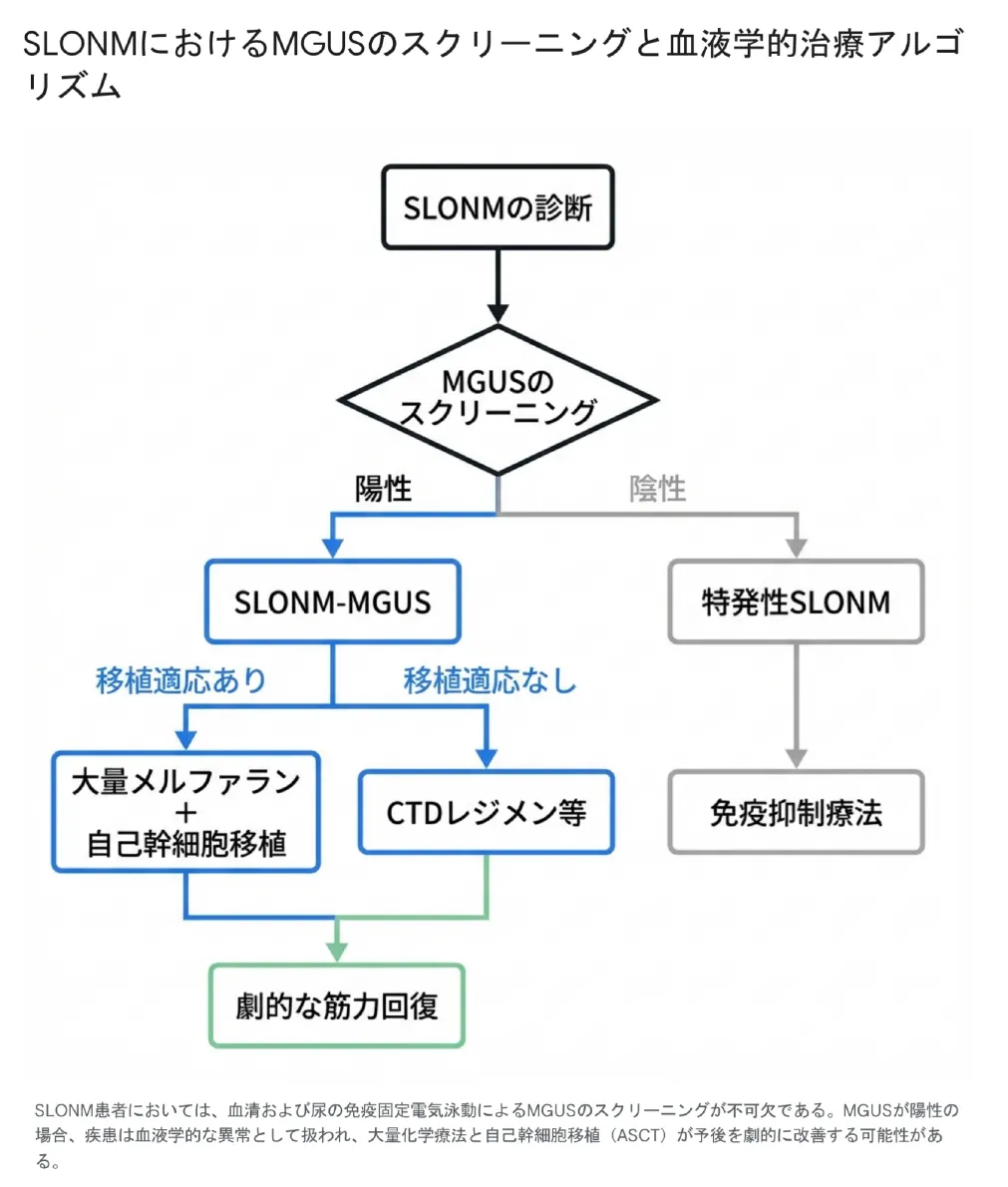
図3:SLONMにおけるMGUSのスクリーニングと血液学的治療アルゴリズム
図の通り、SLONMの診断後はまずMGUSのスクリーニングが必須となります。MGUSが陽性であり、かつ移植適応がある場合、現在最も推奨され、劇的な成功を収めているのが大量メルファラン療法に続く自己幹細胞移植(ASCT)です。この強力な血液学的介入により、車椅子生活だった患者が杖なしで歩行できるようになるなど、驚異的な回復(図の下部の「劇的な筋力回復」)が報告されています。さらに、治療後の筋組織からネマリン小体が完全に消失したことも証明されており、迅速な介入が患者の生死を分ける決定的な要因となります。
6. 現在の標準治療と集学的アプローチ
現在、小児期の遺伝性ネマリンミオパチーに対する根治的な治癒療法は日常臨床には普及していません。そのため、生命予後と生活の質(QOL)を最大化するための多職種による集学的アプローチが中核を成します。
- ➤呼吸器管理(最重要)
四肢の筋力が保たれていても、呼吸機能が先行して悪化することがあります。定期的な肺活量モニタリング、睡眠時の非侵襲的陽圧換気療法(BiPAP)、カフアシストの導入が必須です。 - ➤栄養・消化器管理
球麻痺による嚥下障害のリスクを防ぐため、経管栄養や胃瘻造設術が選択されます。 - ➤整形外科的介入
体幹筋の低下による進行性の脊柱側弯症を防ぐため、装具療法や適切なタイミングでの脊柱固定術が検討されます。
7. 【最新研究】次世代の遺伝子治療と新薬開発の最前線
長らく対症療法に依存してきた医療現場に、病態の根本へ介入する治療法の波が押し寄せています。2025年〜2026年にかけて、前例のない速度で研究が進展しています。
既存薬の再評価(ドラッグ・リパーパシング):サルブタモール
現在最もアクセス可能な薬物治療の有力候補として、喘息治療薬として知られる「サルブタモール(アルブテロール)」が注目されています。骨格筋のタンパク質合成を促進する作用があり、現在欧州において第3相臨床試験(COMPIS試験)が進行中で、ガイドラインを書き換える可能性を秘めています。
AAVベクターを用いた遺伝子治療のブレイクスルー
欠損または変異した遺伝子を直接補完するアデノ随伴ウイルス(AAV)ベースの遺伝子治療が、動物モデルで劇的な成果を上げています。
- ➤ACTA1変異へのハイブリッド療法:異常なタンパク質の生成を強力に抑制しつつ、正常な遺伝子を同時に導入するアプローチが確立されつつあります。
- ➤NEBの巨大遺伝子の壁の突破:遺伝子サイズが大きすぎるネブリンに対し、不可欠な部分だけを残した「ミニ・ネブリン」の設計や、2つのAAVに分割して細胞内で再結合させる「分割AAV療法」が研究されています。
- ➤KLHL40欠損モデルでの生存延長:無治療では生後10日で全滅する重症マウスに対し、最適なAAVを投与した結果、90日を超えて生存し活発に活動するという驚異的な前臨床データが報告されています。
よくある質問(FAQ)
関連記事
参考文献
- [1] Orphanet: Nemaline myopathy. [Orphanet]
- [2] Approach to the diagnosis of congenital myopathies. [PMC]
- [3] Sporadic late-onset nemaline myopathy with MGUS. [Neurology]
- [4] A Foundation Building Strength: Nemaline Myopathy Gene Therapy & Drug Repurposing. [AFBS]
- [5] Changes of Motor Function Tests in Congenital Myopathy Subjects Treated With Oral Salbutamol (COMPIS). [ClinicalTrials.gov]
- [6] Natural History Study for Patients With Nemaline Myopathy in the UK (NatHis-NM-MDUK). [ClinicalTrials.gov]






