目次
- 1 1. TNNT1遺伝子とは:染色体19番が生み出す「遅筋の制御センター」
- 2 2. サルコメアとトロポニン複合体:カルシウムが筋肉を動かすしくみ
- 3 3. 胎児から成熟へ:アイソフォームスイッチングが命運を分ける
- 4 4. 加齢・運動とTNNT1:スプライシングが語る筋肉の適応力
- 5 5. TNNT1変異が引き起こす病気:ネマリンミオパチー5型(NEM5)とは
- 6 6. アーミッシュ・ネマリンミオパチー(ANM):生後18ヶ月という残酷な自然歴
- 7 7. 世界に広がる遺伝的スペクトラム:民族を超えた多様な変異と表現型
- 8 8. 病態メカニズムの解明:「筋力が低下する」のではなく「筋肉が過収縮する」
- 9 9. WiTNNess自然歴研究:臨床試験の礎を築く国際プロジェクト
- 10 10. 次世代の治療フロンティア:遺伝子治療とマバカムテンの可能性
- 11 11. TNNT1の「裏の顔」:複数の悪性腫瘍で機能するオンコジーン的役割
- 12 よくある質問(FAQ)
- 13 関連記事
- 14 参考文献

📍 クイックナビゲーション
TNNT1は、遅筋(タイプ1筋線維)の収縮を制御する「遅筋型トロポニンT(ssTnT)」をコードする遺伝子です。変異が生じると、乳児期に発症し生後数年以内に命を奪う先天性筋疾患「ネマリンミオパチー5型(NEM5)」を引き起こします。一方で近年は、悪性腫瘍の転移・悪性化を駆動するオンコジーン的因子としての顔も明らかになってきました。臨床遺伝専門医が、この多面的な遺伝子の全容を解説します。
Q. TNNT1遺伝子とは何ですか?変異するとどうなりますか?
A. 遅筋の収縮制御に不可欠な遺伝子です。変異によりネマリンミオパチー5型(NEM5)という乳児致死的な先天性筋疾患を引き起こします。近年はAAVを用いた遺伝子治療と、マバカムテンという薬剤による治療研究が急速に進展しています。
- ➤遺伝子の構造 → 染色体19q13.4・14エクソン・遅筋型トロポニンTをコード
- ➤引き起こす病気 → ネマリンミオパチー5型(NEM5)・アーミッシュNM
- ➤病態の核心 → 筋力低下ではなく「筋肉の過収縮」が拘縮を生む
- ➤WiTNNess研究 → 生後60ヶ月での死亡または人工呼吸器管理が100%
- ➤最前線の治療 → マイクロRNAディターゲティング技術を使ったAAV遺伝子治療
1. TNNT1遺伝子とは:染色体19番が生み出す「遅筋の制御センター」
TNNT1(Troponin T1, Slow Skeletal Type)は、ヒトの染色体19q13.4領域に位置する遺伝子で、14のエクソンから構成されています。この遺伝子がコードするのは「遅筋型トロポニンT(slow skeletal Troponin T:ssTnT)」というタンパク質で、262個のアミノ酸残基からなり、分子量は約32.9 kDa、等電点は5.95です。
同じトロポニンT遺伝子ファミリーには、速筋型(TNNT3:染色体11p15.5・19エクソン)と心筋型(TNNT2:染色体1q32・17エクソン)のパラログが存在しますが、TNNT1はイントロンが短く、選択的スプライシングのバリアントも比較的少ないシンプルな構造をしています。長らく「筋肉の構成要素」として基礎研究の範囲で語られてきたこの遺伝子が、今日では希少疾患・遺伝医学・腫瘍生物学の3つの分野で最前線の注目を集めています。
トロポニン(Troponin)は、骨格筋・心筋の収縮を制御する三量体タンパク質複合体の総称です。カルシウムと結合するトロポニンC(TnC)、アクチンに結合してミオシンを抑制するトロポニンI(TnI)、そして複合体全体をトロポミオシンに固定するトロポニンT(TnT)の3種類のサブユニットからなります。TNNT1はこのTnTの「遅筋型」アイソフォームをコードしています。
2. サルコメアとトロポニン複合体:カルシウムが筋肉を動かすしくみ
ssTnTが働く場所は、骨格筋の基本単位であるサルコメア(筋節)です。サルコメアの中で、ssTnTは「細いフィラメント(アクチンフィラメント)」を覆うように伸びる長鎖タンパク質・トロポミオシンに強固に結合し、トロポニン複合体全体をフィラメント上の適切な位置に固定する「アンカー」として機能します。
サルコメアは横紋筋(骨格筋・心筋)の最小機能単位です。太いフィラメント(ミオシン)と細いフィラメント(アクチン)が規則的に配列しており、ミオシンヘッドがアクチンに結合してクロスブリッジ(架橋)を形成することで筋収縮が起こります。トロポニン-トロポミオシン複合体は、このクロスブリッジ形成のカルシウム依存的な「スイッチ」として機能しています。
筋収縮の流れを簡単にまとめると、①筋小胞体からカルシウムイオンが放出される → ②TnCがカルシウムに結合 → ③複合体全体にアロステリックな構造変化が生じる → ④ssTnTを介してトロポミオシンが「横にずれる」 → ⑤ミオシンヘッドとアクチンの結合部位が露出 → ⑥クロスブリッジが形成され筋収縮が起こる、という流れです。ssTnTの構造的完全性は、このカルシウム依存的スイッチを正確に制御するための絶対条件です。
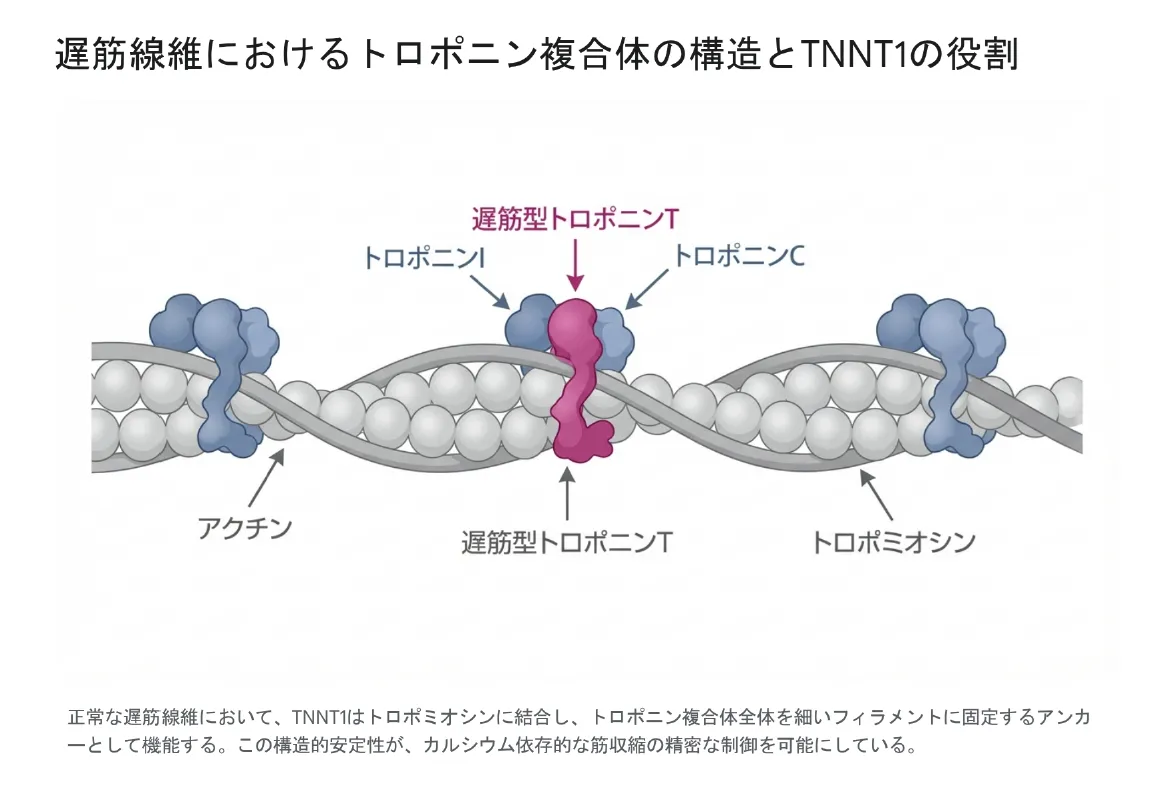
▲ 正常な遅筋線維において、TNNT1(遅筋型トロポニンT)はトロポミオシンに結合し、トロポニン複合体全体を細いフィラメントに固定するアンカーとして機能する。この構造的安定性が、カルシウム依存的な筋収縮の精密な制御を可能にしている。
3. 胎児から成熟へ:アイソフォームスイッチングが命運を分ける
TNNT1遺伝子の発現は、原則として脊椎動物の遅筋線維(タイプ1線維)に厳密に制限されています。しかし胎児の骨格筋では、まだ成熟型のTNNT1は発現しておらず、代わりに胎児型のTNNT3や心筋型のTNNT2が一時的に発現しています。この「代役」が本来の主役に交代するのが、出生後から生後6ヶ月にかけての重要な移行期です。
アイソフォームとは、同じ遺伝子ファミリーに属しながら、異なる組織・発生段階で発現するタンパク質の「型違い」のことです。トロポニンTにはTNNT1(遅筋型)・TNNT2(心筋型)・TNNT3(速筋型)の3つのアイソフォームがあり、発生段階に応じて精密に「スイッチ」されています。このスイッチが正常に行われることが、筋肉の成熟に不可欠です。
このアイソフォームスイッチングのタイムラインが、TNNT1ミオパチーの発症時期と密接にリンクしています。TNNT1変異を持つ新生児は、胎児型アイソフォームが機能している間は比較的症状が軽く見えることがあります。しかし生後数ヶ月で成熟型への置き換わりが進むと、成熟型TNNT1が欠損したまま遅筋線維が機能しなければならない状況になり、急速に症状が顕在化します。「出生時は問題ないように見えた」という経過の謎が、このスイッチングのタイムラインで説明できるのです。
4. 加齢・運動とTNNT1:スプライシングが語る筋肉の適応力
TNNT1は単なる静的な構造タンパク質ではありません。加齢や機械的負荷(運動)に応じて動的に転写・スプライシングが制御される「適応分子」としての側面も持っています。加齢に伴うサルコペニア(筋肉量・筋力の低下)では、速筋線維(タイプ2)から遅筋線維(タイプ1)への移行が進み、TNNT1の発現パターンも変化します。
5ヶ月間の漸進的レジスタンストレーニング(RT)前後でヒト外側広筋を分析した研究では、高齢者の骨格筋においてTNNT1から主に3つのスプライシングパターン(AS1・AS2・AS3)が発現していることが確認されました。慢性的な運動負荷に応答して、AS1は有意に上方制御、AS2・AS3は下方制御されるという劇的な変化が起こります。
📊 AS1/AS2比:筋機能のバイオマーカーとしての可能性
TNNT1のAS2パターンの相対量は、レジスタンストレーニング後の単一筋線維における特異的張力(specific force)と強い負の相関を示します。さらにAS1/AS2比は、トレーニングによる筋線維の張力変化率と明確な正の相関を示しました。この発見は、TNNT1スプライスパターンが機械的過負荷に対するヒト骨格筋の機能的適応を評価する定量バイオマーカーとして機能する可能性を示しています。
5. TNNT1変異が引き起こす病気:ネマリンミオパチー5型(NEM5)とは
TNNT1遺伝子に病原性変異が生じると、「ネマリンミオパチー5型(Nemaline Myopathy type 5:NEM5)」という先天性筋疾患を引き起こします。ネマリンミオパチー全体は新生児5万人に1人以上の割合で発症する非ジストロフィー性の先天性ミオパチーであり、その特徴は筋生検において筋細胞内に「ネマリン小体」と呼ばれる桿状の封入体が観察されることです。
ネマリン小体(nemaline rods)とは、筋細胞の細胞質内に蓄積する「Z帯に類似した高電子密度の桿状封入体」です。ネマリンミオパチーの病理学的特徴であり、筋生検の電子顕微鏡や特殊染色(Gömöri-trichrome染色)で確認されます。NEBやACTA1、TPM3などのサルコメア関連遺伝子が原因となりますが、TNNT1変異によるものは特有の臨床経過をたどります。
NEM5はその大多数が常染色体潜性(劣性)遺伝の形式をとりますが、近年ではドミナントネガティブ効果による常染色体顕性(優性)遺伝の家系も報告されており、遺伝形式の理解は複雑化しています。変異の種類(ナンセンス変異・ミスセンス変異・欠失挿入変異など)によって、症状の重篤度・発症時期・予後が大きく異なります。
6. アーミッシュ・ネマリンミオパチー(ANM):生後18ヶ月という残酷な自然歴
TNNT1関連ミオパチーの中で最も研究が進んでいるのが「アーミッシュ・ネマリンミオパチー(Amish Nemaline Myopathy:ANM)」です。北米のオールドオーダー・アーミッシュのコミュニティでキャリア頻度が約6.5%に達し、TNNT1遺伝子エクソン11のホモ接合性ナンセンス変異(c.505G>T;p.Glu180Ter)を原因とします。この変異では、グルタミン酸のコドンが未成熟な終止コドンに変換され、C末端側の83個のアミノ酸が切り捨てられた短縮型タンパク質が生成されます。
創始者効果とは、小さな集団が大きな集団から分離して独自のコミュニティを形成する際、特定の遺伝子変異が偶然に高頻度で受け継がれる現象です。アーミッシュのような地理的・宗教的に閉鎖されたコミュニティでは、特定のキャリアから集団が始まることでこの効果が顕著に現れ、ANMのようなまれな劣性疾患のキャリア頻度が一般集団の数百倍に達することがあります。
106名のANM患者を対象とした数十年にわたる自然歴研究が、この疾患の容赦ない進行を明らかにしています。患者は平均3.4kgという正常な体重で出生しますが、新生児期から体軸の重度な筋緊張低下(axial hypotonia)、肩や股関節の硬直、下顎・四肢の振戦といった初期症状を呈します。この振戦は生後数ヶ月で収束しますが、その後に近位筋の重篤な拘縮が始まります。
🔴 運動発達の深刻な遅れ
独立して座れた患者は全体のわずか11%。介助なしで立つことは一人もできません。一方で知的能力・受容言語・社会的発達は保たれるという残酷なコントラストがあります。
🫁 胸郭変形と呼吸不全
進行とともに「鳩胸(pectus carinatum)」と呼ばれる重度の胸郭変形が生じ、不可逆的な拘束性肺疾患を引き起こします。反復性の気道感染を経て、全患者が生後6年以内に呼吸不全で死亡します。
ANMの生存期間中央値:18ヶ月(範囲 0.2〜66ヶ月)。生後94%の患者が9ヶ月までに著しい成長障害(Failure to thrive)に陥ります。これは近年まで根本的な治療が存在しない、極めて予後不良な疾患です。

7. 世界に広がる遺伝的スペクトラム:民族を超えた多様な変異と表現型
長らくアーミッシュという特定の遺伝的隔離集団に限定された疾患と考えられてきましたが、次世代シーケンシング技術の普及により、中東・ヨーロッパ・北米の非アーミッシュ集団でも次々と新規TNNT1変異が同定されています。変異の種類によって、発症時期・重篤度・予後が著しく異なります。
| 民族的背景 | 遺伝子変異 | 病原性メカニズム | 臨床的特徴・予後 |
|---|---|---|---|
| アーミッシュ(ANM) | c.505G>T (p.Glu180Ter) |
C末端切断・完全な機能喪失 | 乳児期発症。生存中央値18ヶ月。致死的 |
| パレスチナ系 | c.574_577delinsTAGTGCTGT | フレームシフト・タンパク質欠損 | 乳児期発症。生存中央値70ヶ月(ANMよりやや長い) |
| フレンチ・カナディアン | c.287T>C (p.Leu96Pro) |
部分的な機能障害(ミスセンス) | 小児期発症・極めて緩徐進行。60代まで歩行可能! |
| オランダ・その他 | エクソン8/9欠失・スプライスサイト変異 | 翻訳後分解・TNTタンパク質消失 | 進行性慢性呼吸不全・早期死亡に至る |
| 多世代家系(常染色体顕性) | c.311A>T (p.Glu104Val) |
ドミナントネガティブ機序(常染色体顕性/優性) | 振戦・重度拘縮・小児期発症型と類似 |
特に注目すべきはフレンチ・カナディアンのミスセンス変異(p.Leu96Pro)です。この変異を持つ患者は乳児期致死でなく、「先天性コア・ロッドミオパチー」という全く新しい表現型を示します。筋生検ではネマリン小体だけでなくマルチミニコア(筋原線維内の酸化酵素活性が欠如した微小領域)が観察され、感染症を契機とした横紋筋融解症のエピソードも特徴的です。完全な機能喪失(ナンセンス変異)と部分的な機能異常(ミスセンス変異)の違いが、表現型にこれほど大きな差を生むことを示す好例です。
8. 病態メカニズムの解明:「筋力が低下する」のではなく「筋肉が過収縮する」
TNNT1変異が特定された後も、長年謎だったのは「なぜ筋力低下だけでなく、重篤な関節拘縮・筋硬直という相反するような症状が生じるのか」という問いでした。2024〜2025年にかけて、LaitilaおよびOchalaらを中心とした国際研究チームがこの謎を解明しました。
🔬 細いフィラメントの異常コンプライアンス
正常なssTnTによるトロポミオシンの強固な係留が失われると、アクチンフィラメントの「柔軟性(コンプライアンス)」が異常に増加します。フィラメントが容易に活性化される状態になり、カルシウム感受性が異常に亢進します。
💥 過収縮(Hyper-contractility)
カルシウム感受性の亢進により、筋線維は低いカルシウム濃度でも強く収縮してしまう「過収縮」状態に陥ります。臨床的な関節硬直・拘縮は、この過収縮が直接引き起こしています。
☠️ 短縮タンパク質の細胞毒性
ANM変異では短縮型のssTnT断片が生成されます。この「浮遊断片」がミオフィラメントに組み込まれず、筋細胞内で細胞毒性を発揮し、再生阻害・炎症反応を引き起こす可能性が示されました。
ノックアウト(ssTnT-KO)マウスとノックイン(ANM-KI)マウスを比較した研究では、ANM-KIマウスの方が明らかに重篤なミオパチー表現型を示しました。この結果は、TNNT1関連ミオパチーの病態が単なる「タンパク質の機能喪失」だけでは説明できず、変異断片が積極的に細胞毒性を引き起こす「毒性獲得」のメカニズムも並行して存在することを示しています。
9. WiTNNess自然歴研究:臨床試験の礎を築く国際プロジェクト
将来の遺伝子治療や新薬の有効性を科学的に証明するには、介入なしでの疾患の自然な進行(自然歴)を定量的に把握した「歴史的対照データ」が不可欠です。この目的のため、米国のClinic for Special Children(CSC)が主導し、「WiTNNess」と命名された国際的自然歴観察研究(ClinicalTrials.gov ID: NCT06374719)が実施されました。
2018〜2021年の間に世界中から16名の小児(年齢中央値2.3歳、前向きコホート6名・横断的コホート10名)が登録され、「人工呼吸器非装着生存期間」「成長(Thriving)」「粗大運動機能の獲得」などが厳密に評価されました。その結果は以下のとおりです。
📊 WiTNNess研究:主要臨床マイルストーン達成状況(n=16)
100%
100%
13%
16名中わずか2名のみ
0%
16名中1名も達成せず
出典:WiTNNess: An international natural history study of infantile-onset TNNT1 myopathy(PMC10647004)
また、血液・画像バイオマーカー分析により、TNNT1欠損による一次的な症状発現は骨格筋にのみ限定されており、心筋など他臓器への直接的な損傷は少ないことが確認されました。この知見は遺伝子治療のターゲット設計において重要な意味を持ちます。WiTNNess研究の完遂により、将来の革新的な治験に向けた強固な運用フレームワークが確立されました。
10. 次世代の治療フロンティア:遺伝子治療とマバカムテンの可能性
TNNT1ミオパチーの病理学的特徴の中に、遺伝子治療の成功を強く示唆する事実があります。患者の筋肉では原因タンパク質のTNNT1は欠損していますが、同じ遅筋を構成する遅筋型トロポニンI(TNNI1)やタイプ1ミオシン(MYH7)は依然として発現を維持しているのです。これはタイプ1筋線維自体が死滅しているわけではなく、正常なTNNT1を補充できれば機能回復が見込めることを意味します。
AAV(アデノ随伴ウイルス)ベクターは、病原性のない微小なウイルスを「運び屋」として改変したもので、正常な遺伝子を標的細胞に届ける遺伝子治療の主力ツールです。筋肉への指向性が高いAAV8・AAV9セロタイプが開発されており、スパインラザ(SMA治療薬)やゾルゲンスマなど複数の遺伝性疾患治療薬で実績があります。TNNT1のmRNAは1,239塩基と短く、AAVのパッケージング容量(約4.7kb)に十分収容できます。
マイクロRNAディターゲティング技術:心臓への誤発現を防ぐ精巧な設計
TNNT1遺伝子治療最大の技術的障壁は、野生型TNNT1タンパク質が本来発現すべきでない心筋細胞や速筋線維でAAVによって異所性発現した場合、致死的な心不全を引き起こす危険性でした。この課題を克服するため、マサチューセッツ大学とCSCの共同研究チームが2026年のMDA Clinical & Scientific Conferenceで発表した技術が「マイクロRNAディターゲティング(microRNA de-targeting)」です。
この技術は、AAVベクターの遺伝子発現カセット末端に、心筋・速筋で高発現している内在性マイクロRNAの「標的配列」を組み込むものです。ターゲット組織(遅筋)ではTNNT1が高効率に発現し、非ターゲット組織(心臓・速筋)では内在性マイクロRNAがmRNAを分解・翻訳抑制するというエレガントなメカニズムです。初期実験では非標的組織でのタンパク質発現の全体的な減少が確認され、現在Tnnt1欠損マウスを用いた全身投与による有効性・安全性評価が進行中です。
マバカムテン:過収縮という病態を薬で抑える
マバカムテン(mavacamten)は、ミオシンATPase阻害薬です。本来は閉塞性肥大型心筋症(HCM)の治療薬としてFDAに承認された低分子化合物で、過剰なミオシン-アクチンのクロスブリッジ形成を特異的に阻害し、心筋の過収縮を正常化します。この「収縮を抑える」メカニズムが、TNNT1変異骨格筋の過収縮病態にも応用できることがin vitro機能アッセイで実証されました。
2025年のLaitilaらの研究において、TNNT1変異患者筋線維の異常な過収縮に対し、マバカムテンが細胞レベルで拘縮を可逆的に逆転させうることが示されました。ネマリンミオパチーに対する初の標的薬理学的治療への道を切り開くものとして、極めて高い臨床的意義を持ちます。遺伝子治療の臨床応用が実現するまでの「橋渡し」として、あるいは遺伝子治療との併用として期待されています。
診断の窓(Critical Window)について:高解像度融解解析(HRM法)等により、出生後1〜2日という極めて早期にTNNT1変異を診断することが可能です。不可逆的な骨格変形が進行する前のこの時期が、遺伝子治療を適用するための最重要な治療ウィンドウとなります。
11. TNNT1の「裏の顔」:複数の悪性腫瘍で機能するオンコジーン的役割
筋肉のカルシウム依存的収縮制御という「表の顔」に対し、発癌プロセスおよび腫瘍の悪性進展における役割は、近年発見された驚くべき「裏の顔」です。TCGAやGEO・GTExなどの大規模トランスクリプトームデータベースを用いた解析により、TNNT1遺伝子は正常組織と比較して多くの癌組織で有意に過剰発現しており、患者の予後とも直接的な相関を持つことが確認されました。
上皮間葉転換(Epithelial-Mesenchymal Transition:EMT)とは、癌細胞が原発巣から離脱し他臓器へ転移する際の最重要ステップです。上皮細胞が間葉系細胞の性質を獲得し、細胞間接着が失われ、細胞が遊走・浸潤能力を得る過程をさします。EMTを促進するシグナルとして、Wnt/β-カテニン経路、TGF-β経路などが知られており、TNNT1はこのEMTプロセスを直接駆動することが示されています。
特に大腸腺癌(COAD)、肝細胞癌(LIHC)、膵腺癌(PAAD)、腎淡明細胞癌(KIRC)では、TNNT1の高発現が全生存期間(OS)の有意な短縮と関連しており、独立した予後不良バイオマーカーとして機能しています。肺癌細胞での詳細なメカニズム解析では、TNNT1の過剰発現がWnt/β-カテニン経路を活性化し、癌細胞の移動・浸潤能力を飛躍的に加速させることが示されました。
さらに、KIRC細胞株や肝細胞癌モデルでTNNT1をRNA干渉(shRNA)でノックダウン・中和抗体で機能阻害した結果、癌細胞の増殖・EMT進行が顕著に抑制されました。この事実はTNNT1が単なる「受動的バイオマーカー」でなく、腫瘍の悪性化を直接的に駆動する「能動的な治療標的」である可能性を強く示しています。
よくある質問(FAQ)
🏥 遺伝性疾患・希少疾患の遺伝子検査はミネルバクリニックへ
臨床遺伝専門医が網羅的な遺伝子検査とわかりやすい説明をセットでご提供します。
ダイアモンドプランでは78項目の包括的出生前検査にも対応しています。
関連記事
参考文献
- [1] TNNT1 Gene — GeneCards | TNNT1 Protein | TNNT1 Antibody. [GeneCards]
- [2] TNNT1 — Wikipedia. [Wikipedia]
- [3] Human Slow Troponin T (TNNT1) Pre-mRNA Alternative Splicing Is an Indicator of Skeletal Muscle Response to Resistance Exercise in Older Adults. PMC4296115. [PMC]
- [4] TNNT1 nemaline myopathy: natural history and therapeutic frontier. Human Molecular Genetics. [Oxford Academic] / [PMC]
- [5] Novel autosomal dominant TNNT1 mutation causing nemaline myopathy. PMC5702563. [PMC]
- [6] Slow Skeletal Muscle Troponin T Acts as a Potential Prognostic Biomarker and Therapeutic Target for Hepatocellular Carcinoma. [ResearchGate]
- [7] TNNT1 drives the migration and invasion of lung cancer through Wnt/β-catenin. [ResearchGate]
- [8] Troponin T1 in tumorigenesis and immune modulation: Insights into multiple cancers and kidney renal clear cell carcinoma. PMC11163025. [PMC]
- [9] TNNT1, TNNT2, and TNNT3: Isoform Genes, Regulation, and Structure–Function Relationships. PMC5325693. [PMC]
- [10] Novel Recessive TNNT1 Congenital Core-Rod Myopathy in French Canadians. PMC7078025. [PMC]
- [11] Clinical phenotype and loss of the slow skeletal muscle troponin T in three new patients with recessive TNNT1 nemaline myopathy. PMC8394741. [PMC]
- [12] Pathogenic TNNT1 variants are associated with aberrant thin filament compliance and myofibre hyper-contractility. PubMed 40320982. [PubMed]
- [13] Potential cytotoxicity of truncated slow skeletal muscle troponin T (ssTnT) in a loss of function TNNT1 myopathy mouse model. PMC12354317. [PMC]
- [14] WiTNNess: An international natural history study of infantile-onset TNNT1 myopathy. PMC10647004. [PMC]
- [15] AAV gene therapy for TNNT1-associated Nemaline Myopathy — MDA Clinical & Scientific Conference 2026. [MDA Conference]
- [16] Epigenetic regulation of TNNT1 in gastrointestinal cancers: prognostic implications and clinical significance. PMC12232061. [PMC]
- [17] Latest Update 2025: TNNT1-Related Muscle Weakness — AMB (Accelerated Muscle Biotechnologies). [AMB]
- [18] NCBI Gene: TNNT1 troponin T1, slow skeletal type [Homo sapiens]. [NCBI Gene]






