目次

TPM3遺伝子は、骨格筋の収縮を制御するトロポミオシンをコードする遺伝子です。点変異では先天性ミオパチー、染色体転座では発がんドライバーという、まったく異なる2つの疾患を引き起こす「二面性」を持つ点が、この遺伝子の最大の特徴です。
Q. TPM3遺伝子の変異はどのような病気を引き起こしますか?
A. 大きく2つの方向で疾患を引き起こします。
ひとつは遺伝子内のミスセンス変異による先天性筋線維タイプ不均衡症(CFTD)やネマリンミオパチーなどの先天性ミオパチー。もうひとつは染色体転座によってNTRK1やALKなどのキナーゼ遺伝子と融合し、悪性腫瘍の発がんドライバーへと変貌するケースです。
- ➤TPM3の基本 → 1q21.3に位置し、骨格筋収縮の根幹を担う遺伝子
- ➤先天性ミオパチー → CFTD・ネマリン・キャップ・非定型の4病型と詳細な病態
- ➤発がんメカニズム → TPM3-NTRK1・TPM3-ALK融合とキナーゼの恒常的活性化
- ➤最新の治療 → TRK阻害薬の75%超奏効率、デュアルAAVベクター、CRISPRゲノム編集
- ➤家族歴がある方へ → 遺伝子検査・出生前診断という選択肢の意義
1. TPM3遺伝子とは何か——「二面性」を持つ稀有な疾患関連遺伝子
TPM3(Tropomyosin 3)遺伝子は、ヒト第1染色体長腕(1q21.3)に位置し、細胞骨格の構造維持と骨格筋収縮の根幹を担うアクチン結合タンパク質「トロポミオシン」ファミリーの主要メンバーをコードします。進化的に高度に保存されたこの遺伝子は、胚発生から生後の組織恒常性維持に至るまで不可欠な役割を果たしています。
TPM3遺伝子の最大の特徴は、選択的スプライシングを通じて、低分子量の細胞骨格型アイソフォームと高分子量の横紋筋型アイソフォームという、機能的・空間的に全く異なる複数のタンパク質を生成できる点にあります。さらに、単なる構造タンパク質にとどまらず、ミオシン・トロポニン・コフィリンなどの多様なタンパク質のアクセスを時間的・空間的に制御する「ゲートキーパー」として機能します。
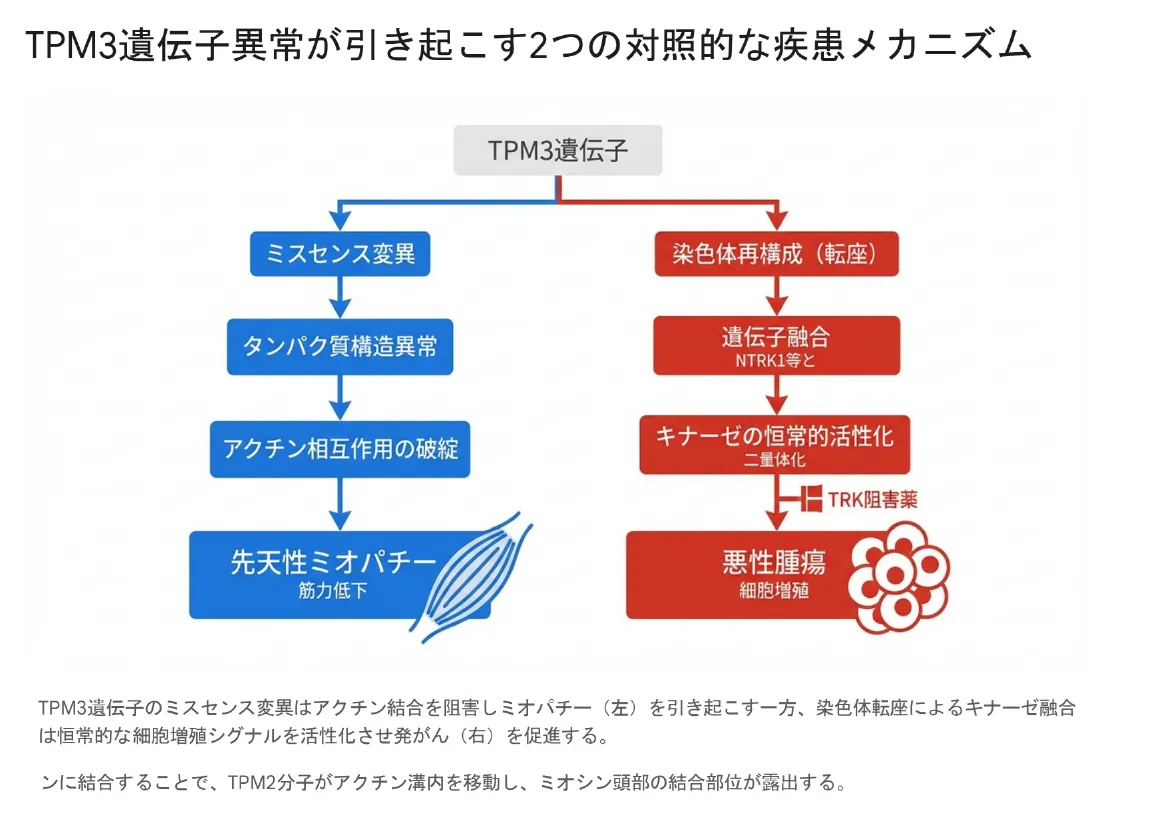
TPM3遺伝子のミスセンス変異はアクチン結合を阻害して先天性ミオパチーを引き起こす一方、染色体転座によるキナーゼ融合は恒常的な細胞増殖シグナルを活性化させ発がんを促進する。
臨床的に特筆すべきは、この遺伝子が持つ「二面性」です。一方では、遺伝子上のミスセンス変異がタンパク質の構造的完全性やアクチンとの結合親和性を損ない、先天性筋線維タイプ不均衡症(CFTD)やネマリンミオパチーといった重篤な先天性ミオパチーを引き起こします。もう一方では、この遺伝子座は染色体再構成(転座)のホットスポットであり、NTRK1やALKといったキナーゼと融合することで、強力な発がん遺伝子(Oncogene)として機能します。ひとつの遺伝子が「筋肉を壊す」方向と「がんを作る」方向、この全く対照的な2つの疾患経路を持つというのは、分子病理学的に極めて稀有な現象です。
筋肉の収縮を「許可」または「禁止」するスイッチタンパク質の一群。アクチンフィラメントに沿って配置され、カルシウムイオン(Ca²⁺)濃度に応じてミオシンの結合をオン・オフします。ヒトには4種類のトロポミオシン遺伝子(TPM1〜4)が存在し、それぞれ異なる組織・細胞で機能します。TPM3遺伝子は特に骨格筋の遅筋線維(タイプI線維)で重要な役割を担います。
2. TPM3タンパク質の構造とアイソフォームの多様性
TPM3遺伝子から生成されるトロポミオシン分子の基本構造は、2本のαヘリックス鎖が互いに巻き付いた「コイルドコイル(Coiled-coil)構造」のホモまたはヘテロ二量体です。これらの二量体はN末端とC末端を介して頭尾結合(Head-to-tail polymerization)することで、アクチンフィラメント全体に沿って途切れのない連続ポリマー鎖を形成します。このポリマーがアクチンの二重らせん構造の溝に沿って配置されることで、細胞の力学的安定性が担保されます。
同じ遺伝子から「選択的スプライシング」によって作られる、構造が少し異なる複数のタンパク質のこと。TPM3遺伝子は骨格筋型の高分子量アイソフォーム(Tpm3.12)と、非筋細胞・細胞骨格型の低分子量アイソフォーム(Tpm3.1など)を生み出します。どのアイソフォームが発現するかは細胞の種類や発生段階によって異なり、それぞれ微妙に異なる機能を担います。
骨格筋型アイソフォーム:Tpm3.12の特殊性
骨格筋において、TPM3遺伝子は「遅筋型αトロポミオシン(Tpm3.12)」と呼ばれる特異的なアイソフォームを提供します。哺乳類の骨格筋には主に姿勢保持・持久力を担うタイプI線維(遅筋)と、瞬発運動を担うタイプII線維(速筋)があり、Tpm3.12はタイプI線維に極めて特異的に局在します。
遅筋線維内ではTpm3.12(γ鎖)がTpm2.2(β鎖)と強固なヘテロ二量体(γβダイマー)を形成します。この組み合わせは速筋線維のαβダイマー(Tpm1.1/Tpm2.2)とは明確に異なる生化学的特性を持ち、遅筋に特有の「ゆっくりだが疲れにくい」収縮特性の分子的基盤となっています。トップダウン質量分析(Top-down MS)を用いた解析でも、ヒト骨格筋ではTpm1.1・Tpm2.2・Tpm3.12の3種が主要アイソフォームとして共存し、その発現比率が筋線維の収縮特性を微細にチューニングしていることが確認されています。
細胞骨格型アイソフォーム:Tpm3.1の多彩な機能
TPM3遺伝子が生み出す低分子量の細胞骨格型アイソフォーム(Tpm3.1など)は、筋肉以外の組織でも極めて重要な役割を果たします。特に注目すべきはインスリン応答性のグルコース取り込みへの関与です。Tpm3.1はGLUT4を含む小胞の細胞膜への輸送と融合の過程において、小胞輸送モーターである非筋ミオシン(Myo2a)の動員を促進する一方、過剰な収縮力を生むMyo1cの結合を制限するという高度な調節を行っています。さらに心筋細胞においては、虚血による酸化ストレスから細胞骨格を安定化させ、心筋保護的な機能も持つことが示されています。
3. 筋収縮を制御する分子メカニズム——「3状態モデル」とデューティ比
骨格筋の収縮は、ミオシン頭部がアクチンフィラメントと相互作用してATPを加水分解する「滑り運動」によって生み出されます。トロポミオシンはこのプロセスにおいて、カルシウムイオン(Ca²⁺)濃度に依存したダイナミックなアロステリック制御スイッチとして機能します。このプロセスは「3状態モデル(Three-State Model)」として概念化されています。
TPM3の変異は、これら3状態間の移行に必要なエネルギー障壁を変化させます。その結果、筋力低下(移行が起こりにくくなる方向)や過収縮(「オフ状態」が維持できなくなる方向)という、全く逆向きの病的表現型が生じます。
ATP加水分解の全サイクルのうち、ミオシンがアクチンに強固に結合して力を発生している時間の割合を指します。Tpm3.12の存在下では、リン酸放出ステップが促進される一方でADP放出が遅延します。その結果、Tpm3.12複合体のデューティ比はTpm1.1(速筋型)複合体の約5倍に増加します。これが、遅筋線維の「収縮速度は比較的遅いが疲労しにくく、持続的な強い張力を維持できる」という生理学的特性を直接生み出しています。

4. TPM3変異が引き起こす先天性ミオパチー——4つの病型と病態
TPM3遺伝子上のミスセンス変異やスプライシング異常は、上述した精巧な分子力学を破綻させ、常染色体優性(顕性)または常染色体劣性(潜性)遺伝形式の先天性ミオパチーを引き起こします。現在までに多数の病原性バリアントが同定されており、変異部位によってアクチンとの結合親和性・トロポニンとの相互作用・コイルドコイル構造の安定性が異なった形で障害されるため、多様な組織学的・臨床的表現型を呈します。
① 先天性筋線維タイプ不均衡症(CFTD)
CFTD(Congenital Fiber-Type Disproportion)とは?
筋生検においてタイプI線維(遅筋)の直径がタイプII線維(速筋)より著しく小さく(通常12〜25%以上の差異)、かつネマリン小体などの他の顕著な病理学的異常を伴わない疾患。現在、TPM3変異はACTA1やSEPN1遺伝子変異を上回り、CFTDの最も一般的な原因として認識されています。
臨床症状の重症度は極めて多様です。乳児期の重度な筋緊張低下(フロッピーインファント)や致死的な呼吸不全から、成人まで気づかれない軽度の筋力低下まで幅広い範囲に及びます。典型的な症状には近位の肢帯筋の筋力低下・頸部屈筋の著明な弱化・足関節背屈筋の弱化・軽度の顔面筋力低下・眼瞼下垂が含まれます。多くの患者は自力歩行を維持しますが、夜間の非侵襲的陽圧換気(NIV)を要する呼吸器合併症を伴うケースが頻繁に観察されます。
分子レベルでは、変異型Tpm3.12が野生型とともにフィラメントに組み込まれ、正常なアクチン・ミオシン相互作用を立体的に阻害する「ドミナント・ネガティブ効果」を通じて収縮力全体を低下させると考えられています。代表的な変異としてはLeu100Met・Arg168Cys・Arg168Gly・Lys169Glu・Arg245Glyなどが挙げられます。
② ネマリンミオパチー(NEM1型)
ネマリン小体(Nemaline bodies / Rods)とは?
筋線維内に形成される異常なタンパク質凝集体。Z帯の構成成分とアクチンから成ります。TPM3変異患者の約29%がネマリンミオパチー(NEM1型)と診断されます。
ネマリン小体形成の背後には、アクチンフィラメントの動態サイクルの崩壊があります。R91PやR91Cといった変異は、トロポミオシンのコイルドコイル構造の熱力学的安定性を著しく低下させます。さらに、これらの変異型Tpm3.12はアクチンフィラメントの切断・脱重合を促進するタンパク質である「コフィリン-2(Cofilin-2)」の機能を強力に阻害します。正常な筋肉の維持には古いアクチンフィラメントの分解と新しいフィラメントの再構築(ターンオーバー)が絶えず必要ですが、このサイクルが滞ることでネマリン小体が沈着していきます。
③ キャップミオパチー(Cap Myopathy)
一部のTPM3変異(R168CやR168Hなど)は「キャップ病(Cap myopathy)」と呼ばれる特異な形態学的変化を引き起こします。筋線維の辺縁部(末梢側)にキャップ状の異常構造物が形成され、ネマリン小体とは異なる染色パターンを示します。変異したアミノ酸がアクチンとミオシンの正常な結合部位に立体的な障害をもたらし、筋収縮を直接妨げることで深刻な筋力低下を招くと推測されています。
④ 新規・非定型表現型(頭蓋顔面異常・過収縮)
近年の全エクソームシーケンシング(WES)の普及により、従来の分類に当てはまらない新たな表現型が次々と明らかになっています。
ひとつは特異な頭蓋顔面異常を伴う症例です。ある15歳男児では、一般的なミオパチー症状に加えて乳児期からの「口呼吸」と「骨格性開咬(Skeletal open bite)」というこれまで報告例のないユニークな表現型が確認されました(新規de novoヘテロ接合性変異 c.44A>G / p.Asp15Gly)。TPM3が顎顔面領域の筋肉の緊張と発達にも関与し、長期的な骨格形成に影響を与える可能性が示されています。
もうひとつは従来の「筋力低下」という常識を覆す「過収縮(Hypercontractile)」の表現型です。p.Glu3Gly変異はトロポニン複合体との相互作用を特異的に不安定化させ、カルシウム感受性を異常に亢進させます。その結果、筋収縮を抑制する「オフ状態」が維持できなくなり、低濃度のカルシウム環境でもアクチン・ミオシン反応が活性化されやすくなります。拘縮や筋緊張亢進といった、通常のミオパチーとは逆向きの臨床症状を呈します。
ひとつの変異したタンパク質が、正常なタンパク質の機能まで積極的に阻害する現象。TPM3変異によるミオパチーの多くでは、変異型Tpm3.12が正常型とともにアクチンフィラメントに組み込まれることで、正常なアクチン・ミオシン相互作用全体を妨害します。これが常染色体優性(顕性)遺伝形式(片方の対立遺伝子に変異があるだけで発症する)の原因となります。
TPM3変異による先天性ミオパチー4病型の比較
| 疾患群 / 病型 | 組織学的特徴 | 主なTPM3変異例 | 主な分子メカニズム |
|---|---|---|---|
| CFTD 先天性筋線維タイプ不均衡症 |
タイプI線維が選択的に低形成(タイプII比12〜25%以上縮小) | Leu100Met、Arg168Cys、Arg168Gly、Arg245Gly | ドミナント・ネガティブ効果による収縮力低下。近位筋優位の弱化・呼吸器障害を伴うことが多い |
| ネマリン ミオパチー NEM1型 |
筋線維内にネマリン小体(Z帯由来成分とアクチンの凝集体)が蓄積 | R91P、R91C、p.Asp15His | コイルドコイル構造の熱力学的不安定化。コフィリン-2阻害によるアクチンターンオーバーの破綻 |
| キャップ ミオパチー |
筋線維辺縁部(末梢側)にキャップ状の異常構造物が形成される | R168H、R168C | アクチン・ミオシン結合の立体的な障害による収縮阻害 |
| 非定型表現型 頭蓋顔面異常/過収縮 |
CFTD類似、または特異な凝集物を伴わない筋線維サイズ異常 | c.44A>G(p.Asp15Gly)、p.Glu3Gly(E3G) | 顎顔面筋の緊張異常による骨格性開咬・口呼吸(p.Asp15Gly)。トロポニン相互作用阻害によるCa²⁺感受性亢進と過収縮・筋緊張亢進(E3G) |
5. 臨床診断のアプローチ——NGS・画像診断・電気生理学
同一の変異(例:p.Arg168His)でも家族内でCFTDとネマリンミオパチーの両方の病理所見を示す事例が報告されるなど、TPM3変異の表現型の多様性は従来の形態学的分類に依存した診断パラダイムの限界を示しています。現在、診断アルゴリズムは筋肉の形態観察からより直接的なゲノム解析へと大きくシフトしています。
遺伝子の塩基配列を大量・高速・低コストで読み解く技術の総称。標的遺伝子パネル検査・全エクソームシーケンシング(WES)・全ゲノムシーケンシング(WGS)などがあります。2020年代の先天性ミオパチー診断では、侵襲的な筋生検の前にNGSを第一選択とすることが国際的に強く推奨されています。乳幼児では全身麻酔を要する生検リスクを回避できる点も大きなメリットです。
NGSの導入は、確定診断までの期間(Diagnostic odyssey)を劇的に短縮するだけでなく、RYR1・NEB・ACTA1などの他の先天性ミオパチー原因遺伝子との鑑別を一度の検査で可能にします。さらに、骨格筋MRIや超音波による筋肉障害パターンの認識が、NGSの標的絞り込みに有力な補助ツールとなっています。TPM3関連ミオパチーでは大腿・下腿において後面より前面の筋群に強い脂肪浸潤・萎縮が現れる特徴的なパターンが報告されています。
また、一部のTPM3変異患者では筋電図(EMG)の単一線維EMGでジッターの増加が観察されたり、重症筋無力症の治療薬(ピリドスチグミン)に陽性反応を示すケースも報告されています。TPM3の機能異常が神経筋接合部(NMJ)の構造的リモデリングをも引き起こす可能性を示唆しており、筋疾患と神経筋伝達障害が重複(Overlap)するという複雑な病態が浮かび上がっています。
6. もう一つの顔——発がんドライバーとしてのTPM3遺伝子融合
TPM3遺伝子の影響は分化した骨格筋の機能不全にとどまりません。がんゲノム分野の研究により、TPM3は染色体再構成(転座や逆位)に関与し、他の遺伝子と融合することで強力な発がん遺伝子(Oncogene)へと変貌することが明らかになっています。
染色体の転座や逆位などの再構成によって、本来別々の2つの遺伝子が結合してひとつのハイブリッドタンパク質を産生する現象。TPM3のコイルドコイルドメインがキナーゼ遺伝子の上流に結合すると、リガンド(細胞外刺激)がなくても自発的に二量体化・自己リン酸化が起こります。これにより細胞増殖・生存シグナル(MAPK・PI3K/AKT・STAT経路など)が無秩序に活性化され、悪性腫瘍の発生と進行が駆動されます。
代表的なTPM3関連遺伝子融合は以下の3つです。
TPM3–NTRK1 融合
大腸がん・甲状腺がん・軟部肉腫・唾液腺がんなどで反復的に同定。NTRK融合は固形がん全体の約0.3%と稀ですが、特定の唾液腺がんや軟部肉腫では比較的高頻度。現在承認済みのTRK阻害薬の主要な治療標的。
TPM3–ALK 融合
炎症性筋線維芽細胞腫(IMT)・未分化大細胞リンパ腫(ALCL)・非小細胞肺がん・腎細胞がんなどの多様な腫瘍カテゴリーで発見。ALKキナーゼの異常活性化を介して増殖シグナルを過剰に駆動。
TPM3–ROS1 融合
非小細胞肺がんなどでALK融合と同様のメカニズムで発がんドライバーとして機能。ROS1キナーゼの恒常的活性化による細胞死の回避と無秩序な増殖を引き起こす。
| 融合遺伝子 | 関連する主な腫瘍タイプ | 発がんの分子メカニズム |
|---|---|---|
| TPM3–NTRK1 | 結腸直腸がん、甲状腺がん、軟部肉腫、唾液腺がんなど | TPM3のコイルドコイルドメインを介したTRKAキナーゼの恒常的二量体化と自己リン酸化による増殖シグナルの暴走 |
| TPM3–ALK | 炎症性筋線維芽細胞腫(IMT)、未分化大細胞リンパ腫(ALCL)、肺がん、腎細胞がん | ALKキナーゼの異常活性化。NTRK1と同様のメカニズムでシグナル伝達を過剰に駆動 |
| TPM3–ROS1 | 非小細胞肺がんなど | ROS1キナーゼの恒常的活性化による細胞死の回避および増殖の促進 |
7. TRK阻害薬によるプレシジョン・メディシンの成功と耐性の課題
TPM3–NTRK1融合をはじめとするNTRK融合遺伝子陽性がんに対する治療は、腫瘍の発生臓器に関わらない「組織非特異的(Histology-agnostic)」な分子標的薬の登場によって、がん医療に劇的なパラダイムシフトをもたらしました。
NTRK融合遺伝子陽性の固形がんに対する分子標的薬。第1世代のラロトレクチニブ(Larotrectinib)とエントレクチニブ(Entrectinib)は、腫瘍の発生臓器に関わらずNTRK融合遺伝子を持つ固形がん患者に対して75%を超える奏効率(ORR)を示し、FDA・EMAから承認を受けています。中枢神経系(CNS)への転移に対しても優れた浸透性を示します。
しかしながら、継続的なTRK阻害薬投与のもとで腫瘍細胞は生存のために二次的な耐性変異(Acquired Resistance)を発生させます。代表的なものが、ソルベント・フロント変異(TRKA G595R)やゲートキーパー変異(F589L)です。これらの変異は薬剤分子がキナーゼのATP結合ポケットに結合することを物理的に妨げ、治療効果を失効させます。
この耐性メカニズムを克服するため、立体障害を回避できる構造を持つ次世代TRK阻害薬(LOXO-195 / セリトレクチニブ、TPX-0005 / レポトレクチニブなど)の開発と臨床試験が現在急ピッチで進められており、がんゲノム医療の最前線となっています。
8. 次世代遺伝子治療の最前線——2026年の到達点
TPM3変異による先天性ミオパチーに対する治療は長らく、理学療法・人工呼吸器による呼吸管理・整形外科的介入といった対症療法に完全に依存してきました。しかし2024年から2026年にかけて、疾患の根本原因に介入する遺伝子基盤治療の研究が前例のない規模とスピードで進展しています。
デュアルAAVベクターアプローチ
AAV(アデノ随伴ウイルス)とは?
遺伝子治療に使われる安全性の高いウイルスベクター。ヒトに病気を引き起こさず、目的の遺伝子を細胞内に効率よく送達できます。デュシェンヌ型筋ジストロフィーなどで臨床試験が進行・承認されており、この技術基盤がTPM3などの希少ミオパチーにも応用されつつあります。
最新の前臨床研究では、単一のAAVベクター内に「疾患の原因を修正する機能置換遺伝子」と「筋肉量を増強するブースター遺伝子」の2種類を組み込む「デュアル遺伝子ベクター(Dual Gene Vector)」アプローチが確立されました。このデュアル遺伝子療法を受けた重篤なマウスモデルでは、すでに進行した筋萎縮状態からでも新たな筋肉量と強度が構築され、歩行機能が完全に正常化(Normalize)するという驚異的な結果が得られています。
アレル特異的サイレンシング(ASO・siRNA)
TPM3ミオパチーの多くは常染色体優性(顕性)遺伝でドミナント・ネガティブ効果を示します。このような疾患では正常な遺伝子を追加するだけでは不十分で、変異アレル(対立遺伝子)から生成される有害なRNAのみを特異的に破壊する「アレル特異的サイレンシング(Allele-specific silencing)」が不可欠です。高度に設計されたアンチセンス・オリゴヌクレオチド(ASO)やsiRNAは単一塩基の違いを識別して変異型mRNAのみを分解でき、次世代の脂質ナノ粒子(LNP)技術による送達効率の飛躍的な改善とともに、臨床開発が加速しています。
CRISPR-CasによるIn Vivoゲノム編集
DNAレベルでの永続的な修復を目指すCRISPR-Cas9などのin vivoゲノム編集技術も、夢の技術から現実の臨床応用へと移行しています。2025年には、個々の患者の固有の遺伝子変異に完全に適応させた完全オーダーメイド(Bespoke)のin vivo CRISPR治療が世界で初めて乳児に対して実施され、その臨床的有効性と安全性が実証されるという歴史的なマイルストーンが達成されました。これにより、TPM3遺伝子上の特定の変異を患者自身の体内で直接DNAレベルで書き換えるという根治的治療への道が明確に開かれつつあります。
📋 自然歴研究(NCT07488806):2026年6月からスペインで開始予定の大規模ネマリンミオパチー患者自然歴研究では、TPM3を含む多様な原因遺伝子を持つ患者コホートを構築し、疾患の進行スピード・呼吸機能低下率・運動機能の経時変化などを長期データ化します。ウェアラブルデバイスによるデジタルバイオマーカー収集も予定されており、将来の遺伝子治療臨床試験のエンドポイント設計に不可欠な基礎データとなります。
よくある質問(FAQ)
🏥 遺伝子のことは、専門医に相談を
TPM3関連疾患の家族歴がある方、遺伝子検査を検討している方へ。
臨床遺伝専門医による正確な情報と、あなたの状況に合った検査の選択肢をご案内します。
参考文献・ガイドライン
- [1] TPM3 Gene – GeneCards. [GeneCards]
- [2] Tropomyosin 3 – Wikipedia. [Wikipedia]
- [3] TPM3: Gene Detail – Cancer Knowledgebase (CKB). [CKB]
- [4] NCBI Gene: TPM3 tropomyosin 3 [Homo sapiens]. [NCBI Gene]
- [5] Marston S, et al. Tropomyosin 3 (TPM3) function in skeletal muscle and in myopathy. J Muscle Res Cell Motil. PMC10629095. [PMC]
- [6] MedlinePlus Genetics: TPM3 gene. [MedlinePlus]
- [7] Fajardo VA, et al. Comprehensive Analysis of Tropomyosin Isoforms in Skeletal Muscles by Top-down Proteomics. PMC4955698. [PMC]
- [8] Tropomyosin 3 Gene Fusions in Cancers: From Mechanisms to Targeted Therapy. PMC12640618. [PMC]
- [9] Structural Effects of Disease-Related Mutations in Actin-Binding Period 3 of Tropomyosin. PMC8622905. [PMC]
- [10] Mutations in TPM3 are a common cause of congenital fiber type disproportion. ResearchGate. [ResearchGate]
- [11] Muscle weakness in TPM3-myopathy is due to reduced Ca²⁺-sensitivity and impaired acto-myosin cross-bridge cycling in slow fibres. Hum Mol Genet. [Oxford Academic]
- [12] A novel pathogenic variant c.44A>G (p.Asp15Gly) in TPM3 causing the phenotype of CMYP4A: A case report. PMC11925118. [PMC]
- [13] A Novel Variant in TPM3 Causing Muscle Weakness and Concomitant Hypercontractile Phenotype. PMC10671854. [PMC]
- [14] Congenital myopathies: revising and revisiting nomenclature and diagnostic guidelines. ENMC. [ENMC]
- [15] Clinical characteristics and treatment patterns of NTRK fusion–positive solid tumors. JMCP. [JMCP]
- [16] Natural History Study for Patients With Nemaline Myopathy in Spain. ClinicalTrials.gov NCT07488806. [ClinicalTrials.gov]
- [17] CRISPR Clinical Trials: A 2025 Update. Innovative Genomics Institute. [IGI]






