目次
- 1 1. CFL2遺伝子の基本情報:染色体位置・ゲノム構造・組織発現
- 2 2. 筋発生における「アイソフォームスイッチ」:エピジェネティクスとmiRNAによる制御
- 3 3. CFL2タンパク質の生化学的機能:「ヌクレオチド状態センサー」としての独自性
- 4 4. CFL2を制御する多重ネットワーク:pH感受性・リン酸化・PI(4,5)P₂
- 5 5. サルコメアにおける相互作用タンパク質ネットワーク
- 6 6. CFL2変異が引き起こす疾患:ネマリンミオパチー7型(NM7)の全貌
- 7 7. 次世代治療の展望:遺伝子治療・ゲノム編集・小分子化合物(2026年)
- 8 よくある質問(FAQ)
- 9 参考文献・ガイドライン
- 10 関連記事

📍 クイックナビゲーション
CFL2遺伝子がコードするコフィリン-2(Cofilin-2)は、骨格筋・心筋のサルコメアに特異的に局在し、アクチンフィラメントの動的なサイクルをナノレベルで精緻に制御する「分子ナノマシン」です。この遺伝子に変異が生じると、先天性ミオパチーであるネマリンミオパチー7型(NM7)を引き起こします。単なる筋肉の構造タンパク質にとどまらず、筋発生プログラムそのものを制御する「能動的なメディエーター」として、近年その重要性が急速に明らかになっています。
- ➤CFL2遺伝子の全体像 → 染色体位置・4エクソン構造・2種類の転写産物(CFL2a/CFL2b)の違い
- ➤アイソフォームスイッチ → 胎生期から成体へ、DNAメチル化・miRNAが司るCFL1→CFL2の切り替え
- ➤独自の生化学的機能 → ATP型・ADP-Pi型・ADP型のアクチンすべてを解体できる「ヌクレオチド状態センサー」
- ➤多重制御メカニズム → pH感受性(His133)・Ser3リン酸化・PI(4,5)P₂によるON/OFF制御の全貌
- ➤ネマリンミオパチー7型(NM7) → 変異ごとに異なる2つの病態経路、拡大する表現型スペクトラム
- ➤次世代治療の展望 → AAV遺伝子治療・miRNAデターゲティング・CRISPR/LNP送達の最前線(2026年)
1. CFL2遺伝子の基本情報:染色体位置・ゲノム構造・組織発現
アクチン脱重合因子(ADF)とコフィリンは、細胞骨格の主要成分であるアクチンフィラメントを切断・脱重合させるタンパク質ファミリーです。哺乳類にはCFL1(非筋型コフィリン)、CFL2(筋型コフィリン)、DSTN(デストリン)の3種類が存在し、それぞれ組織特異的な役割を担います。CFL1は全身の組織に広く発現しますが、CFL2は主に骨格筋・心筋に特異的に強く発現するのが大きな特徴です。
ヒトのCFL2遺伝子は第14染色体長腕(14q12)に位置し(塩基座標:14:34709113–34714823、リバースストランド)、OMIM登録番号は601443です。マウスのオルソログ(Cfl2)は第12染色体に位置します。
CFL2遺伝子は4つのエクソンから構成されており、5’および3’UTR(非翻訳領域)の選択的利用と選択的スプライシングにより、2つの主要な転写産物が生成されます。
CFL2a(転写産物 a)
エクソン1aを利用。骨格筋・心筋以外の様々な組織にも低レベルで広く分布しています。
CFL2b(転写産物 b)
エクソン1bを利用。成熟したヒトの骨格筋・心筋において極めて特異的かつ強力に発現します。
成人組織のRPKM(リード数正規化指標)では、心臓で32.0、膀胱で27.8と高い値が報告されており、骨格筋での発現量は特に際立っています。遺伝子予測スコアはpLI: 0.11、LOEUF: 0.91と算出されており、機能喪失バリアントへの許容度は比較的高い一方、ミスセンスZスコアは1.34とアミノ酸変化への選択圧がかかっていることを示唆しています。
2. 筋発生における「アイソフォームスイッチ」:エピジェネティクスとmiRNAによる制御
💡 エピジェネティクスとは?
DNAの塩基配列自体は変えずに、DNAメチル化やヒストン修飾などによって遺伝子発現のON/OFFを切り替える仕組みです。発育段階に応じて遺伝子発現を柔軟に制御できる「可逆的な遺伝子調節機構」として注目されています。CFL2の発現増加もDNAメチル化の低下(脱メチル化)が主要なトリガーとなっています。
哺乳類の筋肉発達において、正常な筋線維の成熟に不可欠な生理的イベントが「CFL1からCFL2へのアイソフォームスイッチ」です。胎生期の初期骨格筋や幼若な筋芽細胞では、CFL1とCFL2が共発現しており、CFL1が優位に機能しています。しかし発育が進み筋管が形成されるにつれて、CFL1の発現が急速に低下し、CFL2が支配的なアイソフォームとして台頭します。成体の筋組織では、CFL2の相対発現量はCFL1の約55倍にまで達します。
発達段階に伴う筋組織におけるCFL1・CFL2 mRNA発現の転換
(ウシ筋組織モデルより。相対的mRNA発現量の概略値)
CFL1(非筋型)
CFL2(筋特異的)
胎生期(Fetal)
幼少期(Calf)
成体(Adult)
Data source: MDPI Agriculture(ウシ筋芽細胞分化の比較解析より)
DNAメチル化とmiRNAがスイッチを司る
このスイッチを駆動する中心的な機構がDNAメチル化です。胎生期の筋組織では、CFL1・CFL2双方のプロモーター領域のメチル化レベルが比較的高く保たれています。しかし成長とともに、CFL2のプロモーター領域のメチル化が著しく低下(脱メチル化)し、遺伝子発現が一気に活性化されます。この結果として成体の筋ではCFL2のmRNA発現量が胎生期と比べて飛躍的に増大します。
さらに、マイクロRNAの一種であるbta-miR-183がCFL2のmRNAを標的とし、その翻訳を負に制御することで筋芽細胞の分化タイミングを微調整しています。in vitroの初代筋芽細胞実験では、CFL2を過剰発現させると主要な筋分化マーカー(MYOD・MYOG・MYH3)の発現が強力に促進され、逆にCFL2をノックダウンすると筋分化が阻害されることが確認されています。
⚠️ 重要ポイント:CFL2は単なる「細胞骨格の構成部品」ではなく、筋分化シグナルネットワークの中心的なメディエーターとして、遺伝子発現プログラムそのものに能動的に関与しています。
3. CFL2タンパク質の生化学的機能:「ヌクレオチド状態センサー」としての独自性
💡 サルコメア(筋節)とは?
骨格筋・心筋の収縮を担う基本構造単位です。アクチンからなる細いフィラメントとミオシンからなる太いフィラメントが規則的に並び、これらが互いにスライドすることで筋収縮が起こります。CFL2は細いフィラメントの長さを精密にコントロールし、サルコメアの機能を維持しています。
CFL2の最も根源的な生化学的機能は、F-アクチン(線維状アクチン)に特異的に結合し、フィラメントを切断(セバリング)するとともに、ポインテッドエンド(マイナス端)からのアクチン単量体の解離を促進することで、フィラメントの長さを厳密に制御することです。CFL2はCSPR3タンパク質との相互作用によってその脱重合活性がさらに精緻に制御されています。
CFL2の際立った独自性は、CFL1やADF(デストリン)とは明確に異なる、アクチンの「ヌクレオチド状態」に対する特異な親和性にあります。重合直後のF-アクチンはATPを結合しており、時間経過とともにADP-Pi型→ADP型へと変化します。アクチンのW-loop領域(残基165–172)はこのヌクレオチド状態の変化に鋭敏に応答し、各種アクチン結合タンパク質との親和性を決定する「ヌクレオチド・センサー」として機能します。
▼ CFL2タンパク質の活性を調節する多重分子ネットワーク(クリックで拡大)
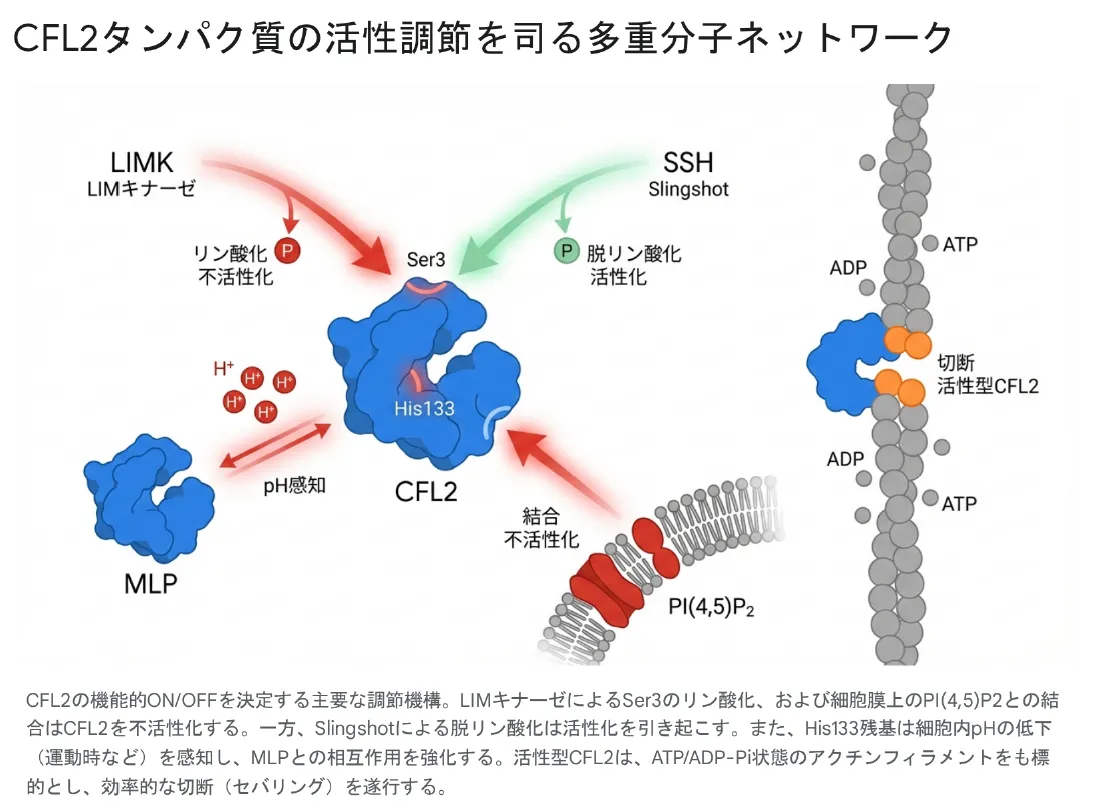
LIMキナーゼによるSer3のリン酸化(不活性化)とSlingshotによるSer3の脱リン酸化(活性化)が中心軸を成す。His133残基はpHセンサーとして機能し、MLPとの相互作用を介して運動時の動態を制御する。活性型CFL2はATP・ADP-Pi・ADP型のアクチンすべてを切断対象とする。
この独自の生化学的プロフィールは、成熟サルコメアのM帯近傍という特殊な環境への進化的適応です。ここではADP型とATP/ADP-Pi型のアクチンサブユニットが複雑に混在しており、CFL2はその状態を問わず一括してフィラメント長を「トリミング」するために、この特異な能力を獲得したと考えられています。
4. CFL2を制御する多重ネットワーク:pH感受性・リン酸化・PI(4,5)P₂
① His133が担う精緻なpH感受性
CFL2の活性は、細胞内pHの変動に対して鋭敏に応答します。この精緻なpH感受性を担う分子基盤が、CFL2の立体構造中に高度に保存されたヒスチジン残基(His133)です。ヒスチジンは生理的pH付近にpKaを持つため、わずかなプロトン濃度の変化によってプロトン化/脱プロトン化が切り替わる、理想的なpHセンサーとして機能します。
in vitroのマイクロ流体アッセイでは、生理的pH(7.0〜7.4)においてCFL2がバーブドエンド(プラス端)から「止まらない(unstoppable)」脱重合を引き起こすことが示されており、この条件下でCFL2がフィラメント脱重合の最大の貢献者となります。
高強度の急性運動時には乳酸蓄積によって細胞内が酸性化します。このときHis133がプロトン化状態となり、MLP(Muscle LIM Protein)との相互作用が著しく強化されます。これは運動負荷に応じてサルコメアのアクチン動態を即座に調節する、極めて合理的な生理的フィードバック機構です。
② Ser3のリン酸化/脱リン酸化による活性のON/OFF
💡 リン酸化(Phosphorylation)とは?
タンパク質のアミノ酸残基(特にセリン・スレオニン・チロシン)にリン酸基が付加される化学修飾。多くのタンパク質の活性スイッチとして機能し、CFL2ではSer3のリン酸化が活性抑制、脱リン酸化が活性回復を意味します。
CFL2の活性制御の中心軸はN末端付近のセリン残基(Ser3)のリン酸化です。Rhoファミリー GTPase(RhoA・Rac1・Cdc42)からの細胞外シグナルが、LIMキナーゼ(LIMK1・LIMK2)やTESキナーゼ(TESK1・TESK2)を活性化し、Ser3をリン酸化することでCFL2のF-アクチン結合と切断活性を完全に抑制します。逆に活性化シグナルが入ると、SlingshotホスファターゼやChronophinがSer3のリン酸基を除去し、CFL2の活性を即座に回復させます。このLIMK–SSH間の絶妙なバランスが、筋細胞の運動・形態変化を規定しています。
③ PI(4,5)P₂との結合による膜上での空間的制御
CFL2は細胞膜上のリン脂質、特にホスファチジルイノシトール4,5-ビスリン酸(PI(4,5)P₂)と多価的・協同的に結合することでも強力に不活性化されます。この結合もHis133を介したpH感受性の影響を受けており、局所的なpH変動と脂質代謝のダイナミクスが協調してCFL2の活性を空間的に規定します。リン酸化された不活性型CFL2は、さらにPLD1(ホスホリパーゼD1)の活性化に関与するという二次的な機能も報告されています。

5. サルコメアにおける相互作用タンパク質ネットワーク
CFL2の機能は、サルコメアを構成する多様なタンパク質との相互作用によってさらに洗練されています。骨格筋の細いフィラメントはアクチン単独ではなく、トロポミオシン(Tpm)・トロポニン(Tn)複合体と結合した状態でカルシウムイオン依存的な収縮制御を受けています。
トロポミオシン(Tpm3.12)
CFL2のF-アクチン親和性自体は低下させないが、フィラメント上への結合協同性を減少させ、切断後の脱重合を抑制することでフィラメントを安定化させる。
Arp2/3複合体
CFL2が切断して生じた新たなポインテッドエンドが、Arp2/3の作用をバイパスしてトロポミオシンのフィラメントへの動員を促進。ラメリポディア様ネットワークを安定したラメラ状フィラメントへ変換する「オーケストレーター」として機能する。
MLP(Muscle LIM Protein)
His133がプロトン化された酸性条件下(運動時)でCFL2と特に強く結合し、サルコメアのアクチン動態を運動負荷に応答させるフィードバック機構を担う。
CAP2 / CSPR3
CAP2はCFL2と相乗的に働き、アクチンアイソフォームの切り替えと脱重合を促進。CSPR3はCFL2と直接会合し、F-アクチン脱重合活性を制御する。
また、筋収縮サイクルとも密接に同期しており、トロポニンからカルシウムが解離した筋弛緩状態ではCFL2のアクチンへの親和性が約2倍に増加することが観察されています。これはCFL2が収縮サイクルと連動して、弛緩期にアクチンのリモデリングを行っていることを示唆しています。
6. CFL2変異が引き起こす疾患:ネマリンミオパチー7型(NM7)の全貌
骨格筋の重篤な筋力低下を特徴とする先天性ミオパチー(生まれつきの筋疾患)の一群です。筋生検でネマリン小体(Nemaline bodies)と呼ばれる棒状の異常構造物が確認されることが診断の決め手となります。ACTA1・NEBをはじめ少なくとも12以上の遺伝子変異が関連することが知られており、CFL2変異によるものが7型(NM7)です。詳細はネマリンミオパチーとは?原因遺伝子・症状から最新の遺伝子治療まで徹底解説もご参照ください。
💡 常染色体劣性(潜性)遺伝とは?
両親から変異遺伝子を1コピーずつ受け継いだとき(ホモ接合体)、または異なる変異を1コピーずつ受け継いだとき(複合ヘテロ接合体)に発症する遺伝形式です。変異を1コピーだけ持つ保因者の両親は通常、症状を持ちません。NM7はこの常染色体劣性(潜性)遺伝形式をとります。
臨床症状と拡大する表現型スペクトラム
典型的な症状は出生時からの重度な筋緊張低下(フロッピーインファント)、著しい運動発達遅滞、頻繁な転倒、歩行・走行の困難です。しかし近年のゲノム解析の進展により、NM7の表現型スペクトラムがはるかに広いことが明らかになっています。
🔴 典型的な先天性発症
出生時から重度の筋緊張低下(フロッピーインファント)、運動発達遅滞、歩行困難。筋生検でネマリン小体が確認される。
🟡 硬直脊椎症候群
小児期(7歳頃)に頸部屈曲不能・進行性脊柱直立化・肩甲骨翼状化を伴う特殊な表現型も報告されている。
🔵 遅発性・成人型
先天性症状は比較的軽度でも、40代以降に急速に進行する遅発性・進行性ミオパチーを呈するケースが確認されており、成人のミオパチー鑑別診断に含めることが提唱されている。
変異型別の分子メカニズム:2つの異なる病態経路
同定されているミスセンス変異(p.A35T・p.Cys39Gly・p.Asp86Asn・p.Lys95Glu)は、それぞれ異なる分子メカニズムで筋障害を引き起こします。
正常なアクチンのターンオーバーが阻害された結果、速やかに分解されるべき古いフィラメントがZ帯近傍などに停滞・蓄積し続け、ネマリン小体や細胞質小体などの巨大タンパク質凝集体を形成して、筋収縮のスライディング機構を破綻させると推測されています。
ノックアウトマウスが示す病態の深刻さ
Cfl2-/-(ノックアウト)マウスでは、発生自体は正常に進行しますが(胎生期はCFL1が補完)、生後早期(P3頃)から重篤な発育不全を示し、生後8日(P8)までに致死となります。筋力低下のため母乳を吸えず、胃が空虚になるのが直接の死因です。組織学的には筋線維の著しい風船様変性(Ballooning degeneration)が広範に見られます。
細胞レベルの解析では、CFL2の欠如が単なるアクチン蓄積のみならず、筋幹細胞による再生修復能の著しい低下・アポトーシス亢進・重度のミトコンドリア機能障害という複合的なカスケードを引き起こすことが示されています。また、ショウジョウバエの病態モデルでも、筋シナプス後部のアクチン組織化の崩壊と神経筋接合部(NMJ)の機能不全が確認されており、CFL2が神経信号の筋肉への伝達においても構造維持に不可欠であることが立証されています。
7. 次世代治療の展望:遺伝子治療・ゲノム編集・小分子化合物(2026年)
💡 アデノ随伴ウイルス(AAV)とは?
遺伝子治療のベクター(運び屋)として最も広く使われるウイルスです。病原性がなく、筋肉・心臓・神経など多くの組織へ効率よく遺伝子を届けられる点が優れています。ただし、患者が保有する中和抗体による免疫原性や、莫大な製造コストが課題です。
2026年現在、NM7を含むネマリンミオパチーに対する承認済みの根本的治療法は存在せず、呼吸管理・理学療法・装具療法などの対症療法が中心です。しかし、世界で約3,200件の遺伝子治療臨床試験が進行中であり、この波は希少筋疾患の領域にも確実に到達しています。
① AAVによる遺伝子補充療法とmiRNAデターゲティング技術
CFL2の機能喪失がNM7の直接原因であることから、正常なCFL2遺伝子をAAVベクターで筋組織に導入する「遺伝子補充療法」が最も有力なアプローチです。ただしCFL2は骨格筋・心筋に強い発現分布を持つため、心筋への過剰発現による心毒性が最大の安全上の課題です。
これを解決するため、心筋・速筋に特異的なmiRNAの標的配列を活用した「miRNAデターゲティング・カセット」をAAVベクターに組み込む高度なエンジニアリングが開発されています。このカセットを認識した内因性miRNAが治療遺伝子のmRNAを選択的に分解し、目的の遅筋線維タイプにのみ安全に治療タンパク質を発現させることが可能です。類縁疾患TNNT1関連ネマリンミオパチー(NEM5)の動物実験でこの技術は有望な成果を収めており、CFL2への応用も強く期待されています。
② 非ウイルス性送達システム(LNP)とCRISPRゲノム編集
AAVの免疫原性・製造コスト問題を根本から解決するため、脂質ナノ粒子(LNP)やウイルス様粒子(VLP)を用いた非ウイルス性送達システムへの移行が急速に進んでいます。将来的には骨格筋の特定受容体を指向する表面修飾を施した次世代LNPを用いて、CRISPR-Cas9などのゲノム編集ツールを直接筋組織へ送達し、患者のCFL2変異塩基を直接修復する「in vivo 遺伝子編集」が主流パラダイムとなることが強く予想されています。
③ 小分子化合物による補助的アプローチ
心筋ミオシン賦活薬のオメカムチブ・メカルビル(Omecamtiv mecarbil)や、リアノジン受容体を安定化させるライカル(Rycals)などの小分子化合物は、カルシウムの過度な流入を起こさずにサルコメアの収縮効率を直接改善するため、補助的治療薬としてのポテンシャルが示唆されています。ただしこれらは、CFL2変異が引き起こす「アクチンの異常蓄積」という根本的な病態の解決にはなりません。遺伝子治療と組み合わせる対症的アプローチとして位置づけられています。
🔬 2026年の動向:AIによる患者の遺伝的背景・エピジェネティクスバイオマーカーの解析を活用し、どのCFL2変異を持つ患者がどの治療法に最もよく反応するかを予測する、真の個別化医療(Holistic patient management)の実現に向けた取り組みが加速しています。
よくある質問(FAQ)
参考文献・ガイドライン
- [1] Cofilin-2 – Wikipedia. [Wikipedia]
- [2] Nakagawa M, et al. Muscle cofilin alters neuromuscular junction postsynaptic development to strengthen functional neurotransmission. Development. 2024;151(13):dev202558. [PMC]
- [3] CFL2 Gene Overview – DECIPHER v11.37. [DECIPHER]
- [4] Functional and Comparative Analysis of Two Subtypes of Cofilin Family on Cattle Myoblasts Differentiation. MDPI Agriculture. 2022;12(9):1420. [MDPI]
- [5] Gonçalves M, et al. Novel missense variants in CFL2 affect F-actin depolymerisation and cause nemaline myopathy. Human Molecular Genetics. 2025;34(17):1471–1484. [Oxford Academic]
- [6] Gurniak CB, et al. Normal myofibrillar development followed by progressive sarcomeric disruption with actin accumulations in a mouse Cfl2 knockout. PMC. 2012. [PMC]
- [7] Kremneva E, et al. Cofilin-2 controls actin filament length in muscle sarcomeres. Developmental Cell. 2014;31(2):215–226. [PMC]
- [8] Xiao L, et al. Magic angle spinning NMR structure of human cofilin-2 assembled on actin filaments reveals isoform-specific conformation and binding mode. PMC. 2022. [PMC]
- [9] New Insight into Muscle-Type Cofilin (CFL2) as an Essential Mediator in Promoting Myogenic Differentiation in Cattle. PMC. 2022. [PMC]
- [10] Demonbreun AR, et al. Congenital myopathies: pathophysiological mechanisms and promising therapies. PMC. 2024. [PMC]
- [11] Muscle LIM Protein interacts with cofilin 2 and regulates F-actin dynamics in cardiac and skeletal muscle. PMC. 2009. [PMC]
- [12] AAV gene therapy for TNNT1-associated Nemaline Myopathy. MDA Conference 2026. [MDA Conference]






