目次

📍 クイックナビゲーション
MYPN遺伝子(ミオパラジン)は、心筋・骨格筋のサルコメアを物理的に支えながら、力学的ストレスを感知して細胞核へシグナルを届ける「二重の役割」を持つ特異な遺伝子です。変異が生じると拡張型・肥大型・拘束型心筋症から先天性ミオパチーまで、多彩な疾患が引き起こされます。このページでは、分子構造から最新の遺伝子治療まで、臨床遺伝専門医が網羅的に解説します。
- ➤MYPN遺伝子・ミオパラジンの基本 → 染色体位置・タンパク質の特徴・ファミリー
- ➤サルコメアZ帯での構造機能 → Ig3ドメインによるアクチン束化と2つの結合サイト
- ➤核内移行とシグナル伝達 → CARP・SRF/MRTF-A経路を介した転写制御の全貌
- ➤心筋症を引き起こす主要変異の比較 → DCM・HCM・RCMを分ける分子メカニズムの違い
- ➤先天性ミオパチーとの関連 → ネマリンミオパチー・垂れ下がり母趾・心筋合併症
- ➤遺伝子治療の最前線 → TreatMYPNプロジェクトとAAVベクター技術の現状
1. MYPN遺伝子とは:サルコメアの「構造の柱」にして「情報の使者」
MYPN遺伝子(Myopalladin;ミオパラジン)は、ヒトゲノムの染色体10q21.3領域に位置し、心筋・骨格筋(横紋筋)に特異的に発現する約145〜147 kDaの大型タンパク質をコードしています。OMIMでの登録番号は *608517 です。
構造の観点から見ると、MYPNはパラジン(PALLD)・ミオチリン(MYOT)と並ぶ「アクチン結合性Igドメイン含有Z帯タンパク質ファミリー」の一員です。特にパラジンとはアミノ酸配列レベルで68%という高い相同性を持ち、どちらも5つの免疫グロブリン(Ig)ドメインとプロリンリッチ領域を備えています。ただし機能面では重要な違いがあり、パラジンがアクチン重合を促進するのに対し、MYPNはアクチンの重合を抑制しながら脱重合も強力に防ぐという独自の制御を行います。
「筋肉(myo)」+「パラジン(palladin)」から名付けられた横紋筋特異的タンパク質。染色体10q21.3にコードされ、サルコメアのZ帯からI帯にかけて局在。単なる構造タンパク質にとどまらず、筋細胞の機械的刺激を化学シグナルへ変換する「メカノセンサー」としても機能する。
MYPNが特筆すべき理由は、その「二重の顔」にあります。一つはサルコメアZ帯を物理的に支える「構造的足場(スキャフォールド)」としての役割。もう一つは筋収縮や機械的ストレスを感知して細胞核へ移行し、遺伝子発現を動的に調節する「多機能メッセンジャー」としての役割です。この二面性ゆえに、MYPN遺伝子の変異は単純な構造破綻だけでなく、細胞内シグナル伝達ネットワーク全体の異常を招き、拡張型心筋症(DCM)・肥大型心筋症(HCM)・拘束型心筋症(RCM)・先天性ミオパチーなど、一つの遺伝子の変異でありながら多彩な疾患表現型を生み出します。
2. サルコメアZ帯での構造的役割:Ig3ドメインとアクチン束化の精密メカニズム
心筋・骨格筋の収縮の最小単位。Z帯(境界線)・I帯・A帯から構成され、アクチン細フィラメント・ミオシン太フィラメント・タイチン・ネブリンなど数十種類のタンパク質が高度に組み合わさった精巧な分子機械。サルコメアが規則正しく並ぶことで筋肉の「横紋(ストライエーション)」が生まれる。
MYPNはサルコメアのZ帯からI帯にかけて局在し、この領域での巨大タンパク質複合体の組み立てと安定性に欠かせない役割を果たしています。具体的には、骨格筋ではネブリンを、心筋ではネブレチンを、それぞれZ帯の主要な架橋タンパク質であるα-アクチニンへ直接連結(テザリング)する役割を担っています。また、MYPNのC末端領域はZ帯内に位置するタイチンのIgドメイン(Z4-Z5領域)にも直接結合し、サルコメア全体の巨大な構造を一体化させています。
Ig3ドメイン:2つの結合サイトによるアクチン束化の仕組み
MYPNの構造的役割の核心をなすのが、タンパク質中央部に位置する第3免疫グロブリン様ドメイン(Ig3ドメイン)です。このドメインは単体でF-アクチン(線維状アクチン)に結合し、複数のアクチンフィラメントを束化(バンドリング)する「最小のアクチン結合モジュール」として機能します。
アラニンスキャン変異導入法などの生化学的解析によって、アクチン結合に不可欠な2つの塩基性アミノ酸クラスターが特定されています。
結合サイト1(Site 1)
K949・R950・K952・R955 の4つの塩基性残基から構成。これらをアラニンに置換すると、F-アクチンへの親和性が著しく低下し、束化能力が完全に消失する。
結合サイト2(Site 2)
K987・R988 の2つの残基から構成。サイト1と独立して機能し、単一のIg3ドメイン上で複数のアクチンフィラメントを同時に捕捉・架橋することを可能にする。
注目すべきは、MYPNのIg3ドメインがオリゴマー(多量体)を形成せず単量体(モノマー)として機能する点です。これは「1分子の上に2つの独立した結合面がある」ことで、複数のアクチンフィラメントを架橋できることを意味します。高密度に多種タンパク質が充填されたZ帯というきわめて狭い空間では、単量体として精密かつコンパクトに機能するこのメカニズムが、空間的制約への最適な進化的適応と考えられています。
3. 核内移行(シャトリング)とシグナル伝達:CARP経路とSRF/MRTF-A経路
MYPNは構造タンパク質でありながら、筋収縮・伸展・疾患による力学的過負荷といった生体力学的ストレスに応じて細胞質から細胞核へと移行(シャトリング)するという、極めて動的な特性を持っています。この機能こそが、MYPNを単なる「筋肉の接着剤」から「遺伝子発現のスイッチ」へと引き上げている最大の理由です。
💡 用語:メカノトランスダクション
「力学的刺激(mechano)→化学シグナル(transduction)への変換」の意。筋肉が受けた物理的な力を細胞内の生化学反応に翻訳するプロセス。MYPNはこの変換装置の中心的な構成要素のひとつ。
以下の図は、MYPNがサルコメアZ帯での構造維持と核内での転写調節という二つの役割を担う様子を示したものです。
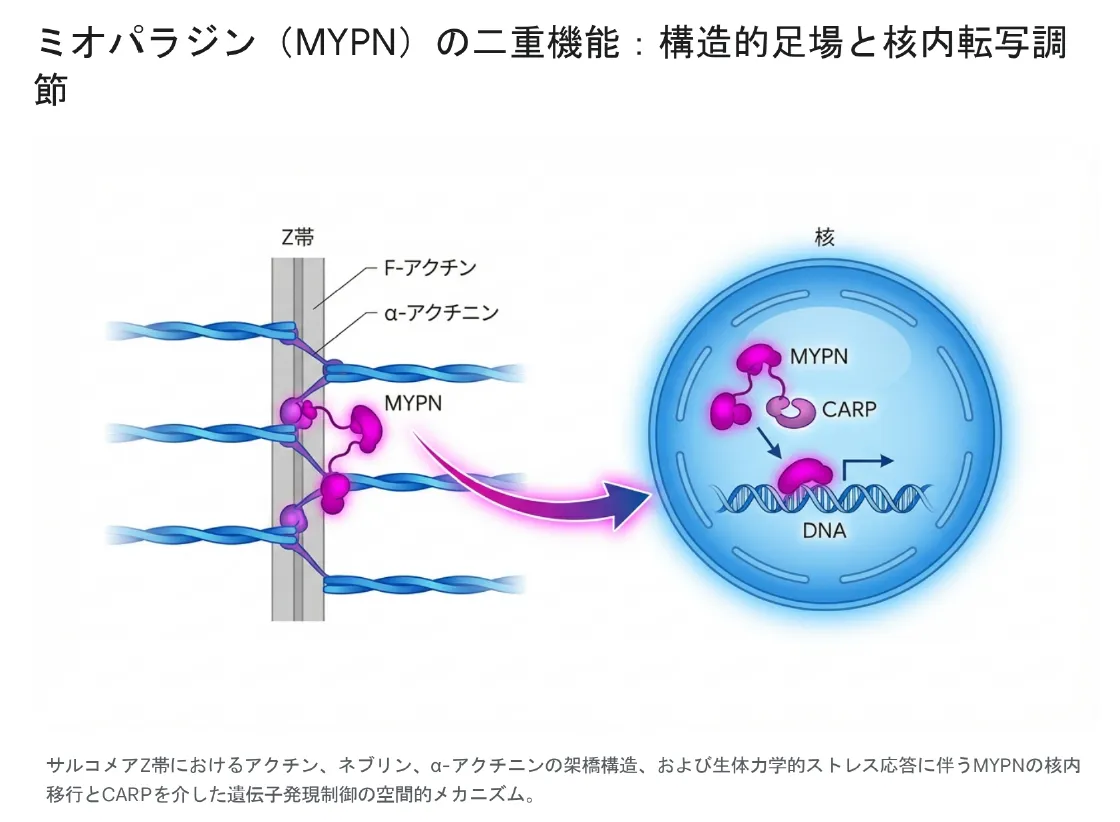
サルコメアZ帯においてF-アクチン・α-アクチニンと結合するMYPN(左)と、生体力学的ストレス応答時にCARPと複合体を形成して核内へ移行し遺伝子発現を制御する様子(右)。
① CARP(心臓アンキリンリピートタンパク質)との相互作用
💡 用語:CARP(Cardiac Ankyrin Repeat Protein)
心筋・骨格筋が虚血・肥大刺激・機械的伸展などのストレスに曝露された際に強力に誘導されるストレス応答性の転写調節因子。Nppa(心房性ナトリウム利尿ペプチド)・Myl2・Tnnc1など、心筋機能に直結する主要遺伝子群の発現を負に制御する。
MYPNのN末端領域はI帯および核内においてCARPと強固な複合体を形成し、Z帯で感知した力学的ストレスを化学シグナルに変換して核へ伝達するメッセンジャーとして機能します。ライブイメージング研究では、このMYPN-CARP複合体の形成を人工的に阻害すると、観察されたすべてのサルコメア構成要素が深刻な構造崩壊をきたすことが確認されており、この経路が筋細胞の恒常性維持に絶対的に必要であることが証明されています。
② SRF/MRTF-A経路:骨格筋の成長を決定づけるスイッチ
MYPNのもう一つの重要なシグナル経路が、SRF(血清応答因子)シグナル伝達経路の制御です。このメカニズムはアクチン動態の巧みな利用に基づいています。
🔄 SRF/MRTF-A経路の作動メカニズム(3ステップ)
- 通常、遊離G-アクチン(球状アクチン)はMRTF-Aに結合し、MRTF-Aを細胞質に留め置く。
- MYPNがF-アクチンに結合してフィラメントの脱重合を強力に阻害すると、細胞質内の遊離G-アクチンのプールが相対的に減少する。
- G-アクチンから解放されたMRTF-Aが核へ移行し、SRFと結合して筋肥大・構造タンパク質合成に関わる標的遺伝子群の転写を促進する。
MYPNを完全に欠損させたノックアウト(MKO)マウスでは、SRF標的遺伝子の発現が網羅的RNAシーケンス解析で顕著に低下していることが確認されています。さらに、MKOマウスの筋芽細胞で観察される筋管細胞の萎縮は、恒常的活性型SRFを導入することで完全に回復(レスキュー)されることが実証されており、MYPNがSRF/MRTF-A経路の上流スイッチとして骨格筋の量的成長を決定づけていることが明らかです。

4. MYPN変異と心筋症:DCM・HCM・RCMを分ける分子メカニズム
MYPN遺伝子の変異は、拡張型心筋症(DCM)・肥大型心筋症(HCM)・拘束型心筋症(RCM)・左室緻密化障害(LVNC)など、多彩な心臓疾患の原因となります。大規模遺伝学的スクリーニングによると、MYPN変異による心筋症の有病率は平均で約1.66%と推計されており、特にヨーロッパ系集団の拡張型心筋症患者では全体の3〜4%でMYPN変異が認められます。
心筋症の遺伝形式は多くの場合常染色体優性(顕性)遺伝です。疾患表現型が多彩な最大の理由は「変異がタンパク質のどのインターフェースを傷つけるか」にあります。以下に代表的な変異を解説します。
① Ig3ドメイン内ミスセンス変異:アクチン束化機能の喪失
心筋症に関連する代表的なミスセンス変異(F954L・R955Q・R955W・P961L・C1002W・R1042C)はいずれもIg3ドメイン内に局在し、F-アクチンへの結合インターフェースを阻害してアクチン束化能力を低下・消失させます。
R955W変異
タンパク質の二次構造自体は野生型に近い状態を維持するが、ショウジョウバエ心筋モデルでの発現解析では、正常なZ帯局在を示さず細胞内で異常なタンパク質凝集(クラスター)を形成する。
P961L変異(重症型)
円二色性(CD)スペクトル解析で、Ig3ドメインが「部分的にアンフォールディングした不安定な構造」へ変化することが判明。非特異的凝集が連鎖的に引き起こされ、重度のサルコメア崩壊へとつながる。
② Q529X変異:ナンセンス変異が引き起こす家族性拘束型心筋症(FRCM)
家族性拘束型心筋症(FRCM)の患者家系から同定されたヘテロ接合型ナンセンス変異 MYPN-Q529X(c.1585C>T) は、特異かつ複雑な発症機序を持ちます。
💡 用語:NMD(ナンセンス変異依存mRNA分解機構)
遺伝子配列の中途に早期終止コドンが生じた際、異常なタンパク質が作られる前にそのmRNAを分解する細胞の品質管理システム。多くのナンセンス変異mRNAはこの機構で排除されるが、Q529X変異mRNAはこれを巧妙に「回避(エスケープ)」する。
Q529X変異mRNAはNMDを回避し、分子量約65 kDaの短縮型(トランケーション)タンパク質を安定的に産生します。マウスノックインモデル(Mypn-Q526X)での研究により、この短縮型タンパク質は核内に異常蓄積して「ポイズンペプチド(毒性ペプチド)」として振る舞うことが判明しました。核内蓄積した変異タンパク質は CARP mRNAレベルの異常亢進、Erk1/2・Smad2・Aktといった生存シグナルのリン酸化低下、進行性の心筋線維化を引き起こし、高致死率を伴う拘束型心筋症をもたらします。
③ Y20C変異:核移行不全と介在板の破壊
DCM・HCM患者から同定された Y20C変異 は、Q529Xとは対照的な「機能喪失・局在異常」メカニズムを示します。N末端領域の構造変化によりCARPへの結合能が著しく低下し、力学的ストレス時でも正常な核移行(シャトリング)が行われません。
Y20C変異を心筋特異的に導入したトランスジェニックマウスでは、顕著な肥大型心筋症の発症とともに、心筋細胞間を機械的・電気的に連結する介在板(intercalated discs)の深刻な破壊が観察されました。デスミン・デスモプラキン・コネキシン43(ギャップ結合)・ビンキュリンといった介在板構成タンパク質群の発現分布に重大な異常が生じ、心筋全体の協調的な収縮能力が損なわれます。
主要変異の比較一覧
| 変異 | 位置 | 関連疾患 | 主な分子メカニズム |
|---|---|---|---|
| R955W | Ig3ドメイン | 心筋症(多様) | 二次構造は保持されるがZ帯で異常凝集を形成し束化能喪失 |
| P961L | Ig3ドメイン | 心筋症(重症型) | 構造がアンフォールディングし熱力学的に不安定化、重度サルコメア崩壊 |
| Q529X | C末端側切断 | 家族性RCM(FRCM) | NMD回避・65 kDa毒性短縮タンパク質が核内蓄積→線維化・シグナル低下 |
| Y20C | N末端領域 | HCM / DCM | CARP結合低下による核移行不全→介在板破壊・結合タンパク質発現異常 |
5. 骨格筋疾患:ネマリンミオパチー・垂れ下がり母趾・ノックアウトマウスの知見
MYPN変異は心筋だけでなく骨格筋にも深刻な影響を及ぼし、先天性ミオパチーと呼ばれる筋疾患を引き起こします。しかも骨格筋疾患と心筋症が高率に合併するという点が、MYPN関連疾患の臨床上の重大な特徴です。
① 先天性ミオパチー24型(CMYO24):旧「ネマリンミオパチー11型」
MYPN変異によるネマリンミオパチーは、かつて「Nemaline myopathy 11 (NEM11)」と呼ばれていましたが、現在OMIMでは 先天性ミオパチー24型(CMYO24;OMIM #617336) としてより広い名称でプライマリ登録されています。遺伝形式は常染色体劣性(潜性)遺伝が多く、ホモ接合型またはコンパウンドヘテロ接合型の機能喪失変異で発症します。
発症頻度は約1:50,000とされ、乳幼児期から筋緊張低下(hypomyotonia)、体幹・四肢の筋力低下を呈します。筋生検では光学顕微鏡・電子顕微鏡下で「ネマリン小体(ロッド状異常構造物)」が筋線維内に多数蓄積していることが病理学的指標となります。
【臨床上の重要な注意点】NEBやACTA1変異によるネマリンミオパチーでは心筋が影響を受けるケースは比較的稀ですが、MYPN変異によるネマリンミオパチーでは、骨格筋の進行性筋力低下に加えて、重篤な拡張型心筋症・心不全を高い確率で合併します。実際に、ネマリンミオパチーと診断された3歳男児が急速に進行するDCMと心不全を発症し、既存の内科的治療に抵抗性を示して難治性心不全で死亡したケースが報告されています。MYPN変異が疑われる場合(特に核内ロッドが観察される場合)は、速やかな心機能評価と遺伝子スクリーニングが強く推奨されます。
② 「垂れ下がり母趾(Hanging Big Toe)」を伴う特異な先天性ミオパチー
近年、MYPN遺伝子のホモ接合型機能喪失変異によって、これまで知られていなかった独特の表現型を示す先天性ミオパチーの家系が報告され注目を集めています。最大の特徴は 「垂れ下がり母趾(Hanging big toe sign)」 です。
症状の特徴
下腿の長母趾伸筋(extensor hallucis longus)に選択的な筋力低下が生じ、足指を上方へ伸展しようとしても親指だけが持ち上がらずに垂れ下がった状態になる視覚的に特徴的な徴候。進行性体幹・四肢筋力低下、脊椎側弯、手指関節拘縮、心筋症合併を伴う。
病理所見の特異性
従来のMYPN関連疾患で見られるネマリン小体(ロッド)やキャップ構造は一切観察されない。電子顕微鏡下でのみ確認できる「Z帯の正方形パターンの崩壊・断片化(Z line fragmentation)」のみが認められる独特の病理像を示す。
③ ノックアウト(MKO)マウスから得られた定量的エビデンス
MYPNを完全に欠損させた構成的ノックアウト(MKO)マウスを用いた研究は、MYPNの骨格筋における絶対的な必要性を個体レベルで実証しています。以下のグラフはその定量的データを示しています。
MYPN欠損が骨格筋成長に与える定量的影響
野生型
MKO
体サイズ(体重)
野生型
MKO
筋線維横断面積(CSA)
MYPNノックアウト(MKO)
MKOマウスは野生型と比較して全体重が約13%小さく(体サイズ87%)、筋線維横断面積(CSA)は約48%という劇的な減少(52%)を示す。これはMYPNがSRFシグナル伝達を介した筋肥大に不可欠であることを裏付けている。
さらに、MKOマウスでは連続的な下り坂走行(筋肉に伸張性収縮を強いる負荷)を与えると運動能力が急速に低下し、Z帯の重度な物理的損傷と筋再生の兆候が広範に確認されます。加えて生後8ヶ月目から進行性のZ帯の肥厚・拡大が生じることも明らかになっており、MYPNが日常的な運動や加齢に伴うダメージからサルコメアを保護するために不可欠であることが個体レベルで示されました。
6. 遺伝子治療の最前線:TreatMYPNプロジェクトとAAVベクターの進化
現在に至るまで、MYPN変異に関連する先天性ミオパチーや心筋症に対して疾患進行を根本から止める根治的治療法は確立されておらず、対症療法や重篤例の心臓移植に頼らざるを得ない状況が続いています。しかし近年、AAV(アデノ随伴ウイルス)を用いた遺伝子治療技術の飛躍的な進歩により、根本的な治癒を目指す革新的アプローチが急速に現実味を帯びてきています。
💡 用語:AAV(アデノ随伴ウイルス)ベクター
病原性を持たない小型のウイルスを改変し、治療用遺伝子を細胞内へ届ける「運搬車」。脊髄性筋萎縮症(SMA)に対するzolgensma(ゾルゲンスマ)がFDA承認を受けるなど、神経筋疾患の遺伝子治療で最も実績のあるベクター系。ただし積載できる遺伝子サイズには約4.7 kbの上限がある。
TreatMYPN(2020〜2025年):first-in-human試験を目指す欧州コンソーシアム
欧州の研究機関を中心に進行中の「TreatMYPN」プロジェクトは、MYPN関連先天性ミオパチーに対するAAV遺伝子治療の確立を目的とした先駆的なトランスレーショナルリサーチです。単に疾患の進行を遅らせるだけでなく、既に低下した筋機能の「回復(リストア)」を実現することを最大の目標に掲げ、迅速な第一相臨床試験(first-in-human試験)への移行を目指して強固な前臨床データを構築中です。
このプロジェクトの中核を支えるのが、以下の2つの高度な前臨床プラットフォームです。
① MKOマウスによる縦断的評価
核の異常内部移行増加・リング状線維形成など、ヒト患者病理に酷似した症状を呈するMKOマウスへカスタマイズAAVベクターを全身投与し、機能回復を縦断的表現型解析および網羅的RNA-Seqで追跡。臨床応用に不可欠な分子バイオマーカーを定義中。
② 生体模倣型3D細胞筋モデル(3DMM)
MKOマウス由来の筋サテライト細胞を特殊ハイドロゲル上で三次元培養し、高度に成熟した立体的筋線維組織を構築。α-アクチニンの横紋形成異常や筋構造の溶解という明確な病理表現型をin vitroで再現し、AAV治療効果をヒト生体内に近い環境で検証可能。
次世代技術:デュアル遺伝子ベクターとプロテイン・ステッチング
AAV遺伝子治療の分野では、MYPNをはじめとする筋疾患の治療に直接恩恵をもたらす技術革新が次々と報告されています。
一つ目は、ネブリン(NEB)遺伝子の治療開発で開発された「プロテイン・ステッチング(protein-stitching)」技術です。超巨大遺伝子を複数の断片に分割して別々のAAVで細胞内へ送達し、細胞内機構を利用して機能的な完全長タンパク質として再構築するこの革新的手法は、MYPNの治療ベクター設計にも直接応用が期待されます。
二つ目は「デュアル遺伝子ベクター技術」です。一つのAAVベクター内に「疾患進行を停止させる遺伝子」と「失われた筋力・筋量を再構築するブースター遺伝子」の2種類を同時に組み込む手法で、GNEミオパチーや肢帯型筋ジストロフィー(LGMD)の動物モデルで、正常な歩行能力の完全回復(ノーマライズ)という劇的な結果が報告されています。既に筋萎縮・心不全が進行した状態でも顕著な機能回復をもたらす道を開く可能性があります。
サルコグリカノパチーの臨床試験では、AAV全身投与によって血清クレアチンキナーゼ(CK)が治療前比90%以上減少するという強力なバイオマーカー改善が示されており、AAVベクターを用いた筋組織への広範かつ持続的なデリバリー技術の安全性・有効性が飛躍的に向上していることを裏付けています。MYPN関連疾患も、長らく「対症療法しか存在しない不治の希少疾患」だった時代から、「遺伝子レベルでの精密な介入が可能な疾患」へと大きなパラダイムシフトを迎えつつあります。
🏥 遺伝性心疾患・筋疾患についてのご相談
ミネルバクリニックは、臨床遺伝専門医が直接対応する専門外来です。
遺伝子検査の内容・結果の意味・家族への影響など、丁寧にご説明します。
よくある質問(FAQ)
参考文献・ガイドライン
- [1] MYPN Gene – Ma’ayan Lab. Harmonizome. [Harmonizome]
- [2] Bang ML et al. Myopalladin knockout mice develop cardiac dilation. eLife. 2020;9:e58313. [eLife]
- [3] Cardiomyopathy-associated and basic residue mutations in myopalladin Ig3 domain. bioRxiv. 2025. [bioRxiv]
- [4] Cardiomyopathy-associated and basic residue mutations in myopalladin Ig3 domain. PMC. 2025. [PMC]
- [5] Leber Y et al. Myopalladin promotes muscle growth through modulation of the serum response factor pathway. PMC. 2020. [PMC]
- [6] Dissection of Z-disc myopalladin gene network in restrictive cardiomyopathy using system genetics approach. PMC. 2017. [PMC]
- [7] Nemaline myopathy with dilated cardiomyopathy and severe heart failure: a case report. PMC. 2021. [PMC]
- [8] Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. PMC. 2012. [PMC]
- [9] Congenital myopathy with hanging big toe due to homozygous myopalladin (MYPN) mutation. PubMed. 2019. [PubMed]
- [10] Congenital myopathy with hanging big toe due to homozygous MYPN mutation. PMC. 2019. [PMC]
- [11] TreatMYPN: Treatment for Myopalladin-related Congenital Myopathy. ANR Project. [ANR]
- [12] Treating neuromuscular diseases: unveiling gene therapy breakthroughs and pioneering future applications. PMC. 2025. [PMC]
- [13] MYPN gene. NCBI Gene Database. [NCBI Gene]
- [14] MYPN – Myopalladin. UniProtKB Q86TC9. [UniProt]






