目次

コフィン・シリス症候群2型(CSS2)は、第1番染色体短腕(1p36.11)に位置するARID1A遺伝子の機能喪失型変異によって引き起こされる、極めて稀な先天異常症候群です。特徴的な粗な顔貌・第5指(小指)の爪の低形成・知的障害に加え、小児期の肝臓がん(肝芽腫)の発症リスクが一般人口より約3万倍高いという独自の腫瘍感受性を伴うため、定期的な腫瘍サーベイランスが不可欠となります。日本では指定難病185に登録されており、医療費助成の対象です。
Q. コフィン・シリス症候群2型(CSS2)とはどのような病気ですか?まず結論だけ知りたいです
A. ARID1A遺伝子の働きが失われることで起こる、生まれつきの多発奇形症候群です。1970年にCoffin医師とSiris医師によって最初に報告されました。粗な顔貌・第5指(小指)の爪の欠損・知的障害・心疾患や摂食障害などの内臓合併症を特徴とし、なかでも小児期の肝芽腫(肝臓がん)リスクが3.6%と高いため、3ヶ月ごとのサーベイランス(早期発見のための検査)が必要です。
- ➤疾患の定義 → OMIM #614607、指定難病185、CSS全体の約5〜7%を占める
- ➤分子メカニズム → クロマチン制御の中心「BAF複合体」の機能不全(ハプロ不全)
- ➤主な症状 → 粗な顔貌・第5指爪欠損・知的障害・心疾患・哺乳不良・脳梁低形成
- ➤腫瘍リスク → 肝芽腫の推定有病率3.6%、AACR基準(1%)を明確に上回る
- ➤成人期の自然歴 → 過体重・脊柱側弯・視力障害が新たな課題として顕在化
1. コフィン・シリス症候群2型(CSS2)とは:疾患の定義と歴史的背景
コフィン・シリス症候群(Coffin-Siris syndrome; CSS)は、1970年に米国のGrange S. Coffin医師とEvelyn Siris医師が、特徴的な顔貌・小指の爪の欠損・重度の知的障害を共有する血縁関係のない3人の女児について報告したのが始まりです。特異な四肢の異常から、長らく「第5指症候群(Fifth digit syndrome)」という別名でも呼ばれてきました。
近年の次世代シーケンス技術の進歩により、CSSは単一の遺伝子が原因ではなく、細胞核内でDNAの読み書きを制御する「BAF複合体」のサブユニット遺伝子群の変異によって起こる疾患群(BAFopathies、BAF異常症)であることが明らかになりました。現在までに少なくとも14の原因遺伝子が同定されており、それぞれCSS1型〜CSS12型などに分類されています。
本記事で扱うコフィン・シリス症候群2型(CSS2)は、第1番染色体短腕(1p36.11)に位置するARID1A遺伝子の常染色体顕性(優性)遺伝形式をとる変異によって引き起こされます。OMIM #614607として登録され、CSS全体の約5〜7%を占めると推定される比較的稀なサブタイプですが、最も頻度の高いCSS1(ARID1B関連、約50〜83%)と比較して重篤な内臓奇形を伴いやすく、特有の腫瘍リスクを伴うという臨床的特徴があります。
💡 用語解説:常染色体顕性(優性)遺伝
「常染色体」は、性染色体(X・Y)以外の染色体のこと。「顕性(優性)」とは、2本ある染色体のうちどちらか片方に変異があるだけで症状が現れる性質を意味します。CSS2では、変異したARID1A遺伝子を1コピー持つだけで発症します。ただし患者さんのほとんどは、両親には変異がなく、本人で初めて生じた新生突然変異(de novo変異)によって発症します。これは患者さんの生殖能力が低いため、変異が世代を超えて受け継がれにくいことが理由です。
日本国内において、コフィン・シリス症候群は厚生労働省が定める指定難病(告示番号185)に登録されています。一定の診断基準と重症度基準を満たす患者さんは、医療費助成制度の対象となります。患者数は国内で100人未満と推定される極めて稀な疾患です。
2. 原因遺伝子ARID1Aと分子病態メカニズム
CSS2の理解には、ARID1A遺伝子がコードするタンパク質と、それが構成する巨大な分子マシン「BAF複合体」の働きを知ることが鍵になります。
💡 用語解説:BAF複合体(mSWI/SNF複合体)とは
細胞核の中でDNAは「ヒストン」というタンパク質に巻きついて、ヌクレオソームと呼ばれる構造をつくっています。このDNA+ヒストンの集合体が「クロマチン」です。BAF複合体は、ATPのエネルギーを使ってヌクレオソームを動かし、必要な遺伝子を読み出せる状態(開いたクロマチン)にする巨大なタンパク質マシンです。胚発生・神経発生・心臓の形成など、命の設計図を読み出すあらゆる場面で中心的な役割を果たします。
ARID1A遺伝子は、BAF複合体の中でDNAに結合する「カギ」の役割を担うBAF250a(別名p270)というタンパク質をコードしています。20個のエクソンから構成され、生み出される最長のタンパク質は2,285個のアミノ酸からなる、極めて大型の分子です。タンパク質の中央部にはDNAのATリッチ配列を特異的に認識する「ARIDドメイン」を持ち、これが複合体全体を標的の遺伝子へと案内する司令塔として機能します。
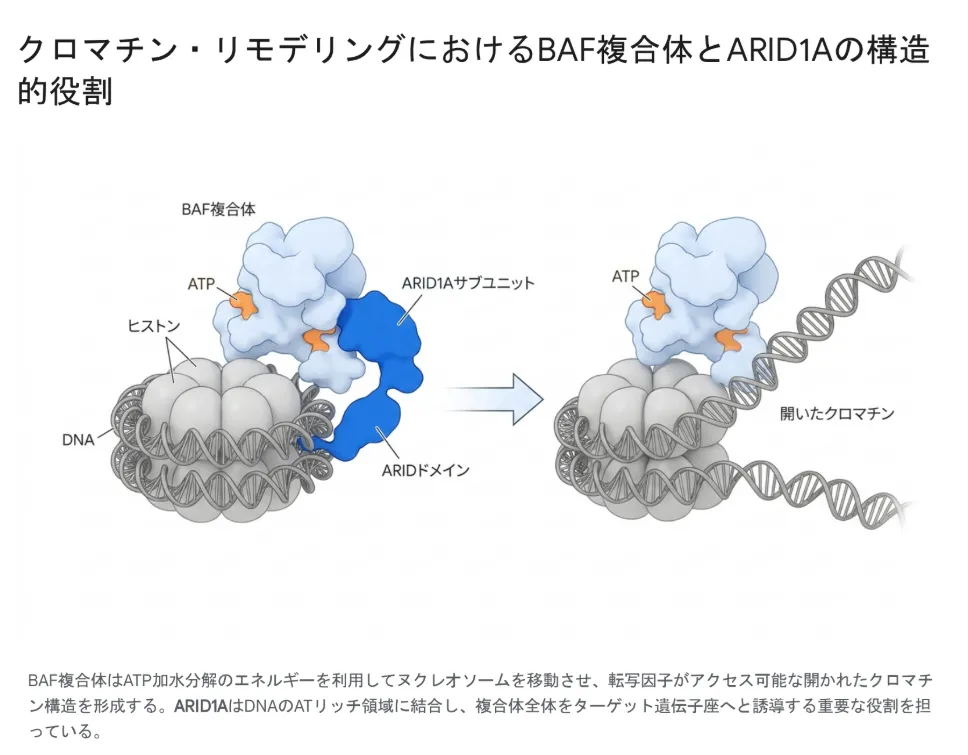
BAF複合体はATPのエネルギーを使ってヌクレオソームを移動させ、遺伝子が読み出せる「開いたクロマチン」をつくる。ARID1AはDNAのATリッチ領域に結合し、複合体全体を標的遺伝子へ誘導する重要な役割を担う。
CSS2の中核メカニズム「ハプロ不全」
CSS2でみられるARID1A変異の大部分は、フレームシフト変異・ナンセンス変異・スプライシング異常を伴う欠失といった、タンパク質の機能を完全に失わせる機能喪失型変異(Loss-of-Function変異)です。これらの変異から生まれた短いmRNAは細胞内の品質管理機構によってすぐに分解され、結果としてARID1Aタンパク質の量が正常の半分以下に低下します。この状態をハプロ不全と呼びます。
💡 用語解説:ハプロ不全(Haploinsufficiency)
遺伝子は通常、父方・母方の染色体に1コピーずつ、合計2コピー存在します。「ハプロ不全」とは、そのうち片方の遺伝子が壊れて働かなくなり、残った正常な1コピーだけでは細胞や体の正常な発生・機能を維持するのに不十分な状態を指します。ARID1Aのように、発生過程で大量に必要とされる遺伝子は、1コピーだけでは「量が足りない」という現象が起こり、結果として複数の臓器に影響が及びます。
💡 用語解説:ミスセンス変異とフレームシフト変異
ミスセンス変異:DNAの塩基が1つ変わることで、タンパク質を作る「設計図」のうちアミノ酸1個が別のアミノ酸に置き換わる変異です。タンパク質の形や働きに微妙〜大きな影響を与えます。
フレームシフト変異:DNAから1〜数個の塩基が抜けたり追加されたりすることで、設計図の「読み枠」全体がずれてしまう変異です。それ以降のアミノ酸配列がすべて狂い、ほとんどの場合タンパク質が完全に機能を失います。
ナンセンス変異:途中に「ストップ命令」が新たに生じる変異で、短いタンパク質しか作られなくなります。
マウスでは胎生致死、ヒトでは生存する不思議:体細胞モザイク現象
マウスを使った基礎研究では、ARID1A遺伝子の片方をノックアウト(破壊)するだけで胎生致死(胚の段階で死んでしまう)になることが報告されています。それにもかかわらず、ヒトのCSS2患者さんが生まれて生存している理由として、体細胞モザイク(Somatic mosaicism)の関与が強く示唆されています。
💡 用語解説:体細胞モザイク
受精後の細胞分裂のごく初期段階で新たな変異が生じると、変異を持つ細胞と持たない細胞が同じ体の中に混在する状態になります。これを「体細胞モザイク」といいます。CSS2患者さんでは、すべての細胞ではなく一部の細胞だけが変異を持つことで、致死的な発生段階を乗り越えて生まれてくることができたと考えられています。モザイクの割合や分布によって、症状の重さや臓器ごとの違いが説明できる可能性があります。
3. 主な症状と表現型スペクトラム
CSS2の症状は多岐にわたります。軽度の知的障害にとどまる稀な症例から、重篤な内臓合併症によって乳幼児期から命の危機に瀕する重症例まで、表現型のスペクトラムは広範囲です。以下、臓器別に主な特徴をまとめます。
👤 顔貌・体毛
- 粗な顔貌(Coarse facies)
- 頭髪の疎毛、低い前頭部生え際
- 濃く太い眉毛・異常に長い睫毛
- 背部や四肢の多毛症:約95%
- 広い鼻尖、厚い鼻翼、前屈した鼻孔
- 広い口・厚く外反した上下口唇
🖐️ 四肢・骨格
- 第5指(小指)末節骨・爪の低形成または欠損:65〜80%
- 第5趾の爪の低形成
- 他の指趾爪の低形成:約40〜50%
- 関節弛緩
- 脊柱側弯症:約40%、成長と共に進行
- 骨年齢の遅延
🧠 神経・発達
- 発達遅滞・知的障害:約98%(中等度〜重度が多い)
- 言語表出の遅れ(約1/3は無発語)
- 中枢性筋緊張低下:約75%
- てんかん発作:約50%
- 脳梁低形成・無形成
- 小頭症・大脳皮質形成異常
❤️ 内臓・全身
- 哺乳不良・体重増加不良:約90%
- 胃食道逆流・反復性嘔吐
- 先天性心疾患(VSD・PDA等):30〜67%
- 反復性気道感染症:約60%
- 難聴:約45%
- 泌尿生殖器の奇形(重複腎盂尿管等)
💡 用語解説:粗な顔貌(Coarse facies)
「粗な」というのは医学的な専門用語で、顔の輪郭や各パーツが大きく、はっきりとした印象を与える顔つきを意味します。具体的には、厚い口唇・広い鼻翼・濃く太い眉毛・低い前頭部生え際などの特徴が組み合わさることで、年齢を重ねるにつれて徐々に明瞭化します。乳児期にはわかりにくくても、幼児期・学童期にかけて特徴が際立っていく傾向があります。決して見た目を否定的に表現する言葉ではなく、医学的な記述用語として用いられます。
💡 用語解説:脳梁低形成・無形成
「脳梁」とは、左右の大脳半球をつなぐ太い神経線維の束で、左右の脳の情報のやりとりを担う重要な橋渡し役です。CSS2では、この脳梁が小さい(低形成)または全くない(無形成)状態がもっとも多くみられる脳の構造異常です。脳MRI検査で評価され、知的障害や運動発達の遅れと関連します。

CSS2はCSS1よりも内臓合併症が重篤になりやすい
CSS2(ARID1A関連)は、もっとも頻度の高いCSS1(ARID1B関連)と比較して、重篤な内臓奇形の合併頻度が高いことが知られています。特に乳児期の哺乳不良は約90%とほぼ全例にみられ、25〜50%の乳幼児が経管栄養や胃瘻造設を必要とするレベルです。先天性心疾患の合併頻度(30〜67%)も他のサブタイプより高く、外科的修復が必要となるケースが少なくありません。
4. 鑑別診断:似ている疾患との見分け方
CSSは遺伝的に多様な疾患群であり、複数の遺伝子変異が類似した臨床症状を示します。さらに、CSSと一部の特徴が重なる別の症候群もあります。正しい治療・サーベイランス計画のため、これらとの鑑別が極めて重要です。
CSSの主要サブタイプ間の比較
| サブタイプ | 原因遺伝子 | 重症度の傾向 | 特有の腫瘍リスク |
|---|---|---|---|
| CSS2 | ARID1A | 中等度〜最重度・内臓合併症が多い | 肝芽腫(3.6%) |
| CSS1 | ARID1B | 比較的軽度〜中等度 | 明確な上昇は確認されていない |
| CSS3 | SMARCB1 | 最重度・言語発達遅滞顕著 | 神経鞘腫症 |
| CSS4 | SMARCA4 | 中等度〜重度・行動異常 | 研究中(ラブドイド腫瘍関連) |
鑑別が必要な類似疾患
ニコライデス・バライツァー症候群
SMARCA2遺伝子のミスセンス変異等が原因で、粗な顔貌・疎な頭髪・重度の知的障害・てんかんなどCSSと酷似します。
鑑別ポイント:指節間関節の隆起が目立つ一方、CSSの決定的特徴である第5指の爪・末節骨低形成がないことが重要な区別点です。
コルネリア・デランゲ症候群
NIPBL等のコヒーシン複合体遺伝子の変異による疾患で、重度の成長障害・多毛症・上肢の還元奇形を呈し、四肢所見がCSSと類似します。
鑑別ポイント:癒合眉(synophrys)・長い睫毛・薄い上唇・下向きの口角といった独特の顔貌、上肢の還元奇形(指の欠損など)で区別されます。
ボージェソン・フォルスマン・レーマン症候群
PHF6遺伝子のX連鎖性変異による疾患で、てんかん・内分泌異常・特徴的な肉付きの良い顔貌・大きな耳介・重度の知的障害を伴います。
鑑別ポイント:遺伝子パネル検査による分子学的鑑別が確実です。
マブリー症候群
PIGV遺伝子等によるGPIアンカー生合成異常症。知的障害・難治性てんかん・粗な顔貌・第5指末節骨低形成を呈します。
鑑別ポイント:血清アルカリホスファターゼ(ALP)の著明な上昇が特徴で、生化学検査が手がかりとなります。
5. 診断の進め方:出生前診断と出生後診断
CSS2を含むコフィン・シリス症候群の確定診断は、臨床所見と分子遺伝学的検査の両方を組み合わせて行います。診断は「出生前」と「出生後」で大きく方法が異なります。混同を避けるためにそれぞれ説明します。
出生後の診断:臨床所見+遺伝子パネル検査
出生後にCSSが疑われる場合、まず以下のような特徴的所見の組み合わせを評価します。
💡 CSS2を疑う主要所見の組み合わせ
- ➤第5指(小指)の爪または末節骨の低形成・欠損
- ➤粗な顔貌(厚い口唇・濃い眉毛・長い睫毛)
- ➤発達遅滞・知的障害・筋緊張低下
- ➤体毛の多毛症・頭髪の疎毛
- ➤哺乳不良・先天性心疾患・脳梁低形成
これらの所見からCSSが疑われる場合、次に分子遺伝学的検査による確定診断を行います。臨床的に最も実用的なのは、コフィン・シリス症候群NGSパネル検査で、ARID1A・ARID1B・SMARCA2・SMARCA4・SMARCB1・SMARCE1・SOX11・PHF6など、CSSの主要原因遺伝子11種類をまとめて解析できます。診断が困難なケースでは、クリニカルエクソーム検査でより網羅的に解析することも可能です。知的障害が前面に出ているケースでは知的障害遺伝子パネル検査からのアプローチも選択肢になります。
💡 用語解説:NGSパネル検査・全エクソーム解析
NGS(Next Generation Sequencing:次世代シーケンサー):DNAの配列を高速で大量に読み取る装置です。多数の遺伝子を同時に解析できるため、似た症状を引き起こす複数の遺伝子をまとめて調べる「パネル検査」が可能になりました。
全エクソーム解析(WES):遺伝子のタンパク質を作る部分(エクソン)全体をまとめて解析する方法。原因遺伝子の絞り込みが難しい複雑な症例で力を発揮します。
出生前診断:羊水検査・絨毛検査による確定診断
CSS2は基本的には新生突然変異(de novo変異)として発症するため、家族内の同じ変異が既に同定されているなど、特殊な状況がなければ出生前に診断することはまずありません。ただし、患者さん本人が将来子どもを持つ場合や、既に家系内に変異が確認されている場合は、絨毛検査または羊水検査による出生前遺伝子診断が選択肢となります。
重要:CSSは表現型が極めて広く、生まれてみないとどの程度の症状になるかが予測できない疾患です。出生前に変異を見つけたとしても、症状の重症度はわかりません。出生前診断を考える場合は、必ず遺伝カウンセリングを通じて、ご家族で十分に話し合ったうえで決定してください。
6. 肝芽腫リスクと腫瘍サーベイランス:CSS2最大の臨床課題
CSS2をARID1B関連CSS1など他のサブタイプと根本的に区別する最重要ポイントが、小児期の肝臓がん「肝芽腫(Hepatoblastoma)」の発症リスクが有意に高いことです。
ARID1A遺伝子は、卵巣明細胞癌・子宮内膜癌・胃癌・大腸癌・肝細胞癌など、ヒトの多くのがんで体細胞変異が頻発する強力な「がん抑制遺伝子」として知られています。CSS2の患者さんは、もともと全身のすべての細胞で片方のARID1A遺伝子が壊れた状態(生殖細胞系列変異)で生まれてきます。そのため、もう片方のARID1Aに体細胞変異が加わると、がん化のブレーキが完全に外れて腫瘍が発生しやすくなる——これが「ツーヒット仮説」と呼ばれる発がん理論です。
ARID1A関連CSS2における肝芽腫の発症リスク比較
3.6%
1.0%
約0.0001%
ARID1A関連CSS2患者の肝芽腫推定有病率3.6%は、米国がん学会(AACR)が定める小児がんサーベイランス推奨閾値1.0%を明確に上回ります。
この数値は単なる偶然では説明できない有意な発症リスクの上昇であり、すべてのARID1A病的バリアントを持つ乳幼児に対して、肝芽腫の早期発見を目的とした積極的なサーベイランスが標準的に推奨されます。
推奨される腫瘍サーベイランスプロトコル
🩸 血清AFP測定
少なくとも3ヶ月ごとに、血清アルファフェトプロテイン(AFP)濃度を測定します。AFPは肝芽腫の最も信頼性の高い腫瘍マーカーです。
🔬 腹部超音波検査
専門医による腹部超音波(エコー)検査を、同じく3ヶ月ごとに実施します。肝臓内の腫瘤性病変を画像でスクリーニングします。
このサーベイランスは、肝芽腫の好発年齢である乳幼児期から幼児期を通じて継続することが推奨されます。なお、ARID1A以外の他のCSS関連遺伝子(ARID1B、ARID2など)では発がんリスクの有意な上昇は確認されておらず、これらのサブタイプにはルーチンの全身がんスクリーニングは推奨されません。例外としてSMARCB1変異例(CSS3)では神経鞘腫症のリスク上昇があり、別の画像評価が必要です。
臓器別の継続的マネジメント
CSS2には根治的な治療法は存在しません。治療の主眼は、症状の緩和と機能発達の最大化、合併症による生命リスクの最小化に置かれます。小児科・臨床遺伝科・リハビリ科・消化器科・循環器科・整形外科・眼科など多職種からなる集学的ケアチームの構築が不可欠です。
栄養・消化器管理
乳児期の哺乳不良・嚥下障害は最優先で対処すべき課題です。経口摂取で必要カロリーが維持できない場合、経鼻胃管栄養を導入し、長期化が予想される場合は胃瘻造設を検討します。
発達支援
理学療法(PT)・作業療法(OT)・言語聴覚療法(ST)を組み合わせた早期療育プログラムが必須です。代替的コミュニケーション手段の導入も重要です。
心臓・呼吸器管理
診断時に必ず心エコー検査を実施し、心疾患の有無を確認します。喉頭軟化症による呼吸障害には適切な気道管理が求められます。
整形外科・感覚器
学童期以降に進行しうる脊柱側弯症への定期的X線評価、装具療法を計画します。聴力・視力検査も定期的に必要です。
7. 成人期の自然歴:CSSは「生涯の疾患」
CSSに関する過去の医学報告のほとんどは小児期の患者を対象としたものでした。小児期を脱した後、患者さんがどのような経過をたどるのかは長らくデータの空白地帯でしたが、2024年にSchmetzらによって発表された分子診断確定済みの成人CSS患者35名を対象とした国際的多施設研究により、成人期の表現型の全容がはじめて明らかになりました。
この画期的な研究で示されたのは、CSSが小児期に特有の発生異常にとどまらず、加齢に伴って表現型が動的に変化する「生涯の疾患」であるという事実です。
| 健康課題の領域 | 小児期の典型 | 成人期の変化 |
|---|---|---|
| 栄養・代謝 | 哺乳不良・体重増加不良 | 過体重・肥満への急速な移行 |
| 筋骨格系 | 関節弛緩・筋緊張低下 | 脊柱側弯症の重症化・有病率上昇 |
| 感覚器系 | 軽度な斜視・眼瞼下垂 | 視力障害の頻度が想定以上に高い |
| 精神・行動 | 発達遅滞・多動 | ASD様・ADHD様の行動異常が顕著 |
これらの新しい知見は、CSS2患者さんのケアが小児科医の手を離れた後も、内科・整形外科・眼科・精神科を含む多職種連携による、生涯にわたるトランジションケア(移行期医療)が不可欠であることを強く示しています。
8. 遺伝カウンセリングと社会的支援
CSS2の診断確定後、ご家族には丁寧な遺伝カウンセリングが不可欠です。臨床遺伝専門医が中立的な立場で、以下のような内容を扱います。
- ➤遺伝形式と再発率:CSS2のほとんどは新生突然変異であり、両親には変異がなく次子の発症リスクは一般人口と同程度です。ただし生殖細胞モザイクの可能性もあるため、絶対0%とは言えません。患者本人が将来子どもを持つ場合は理論上50%の遺伝確率です。
- ➤予後情報の共有:適切な集学的ケアによって、多くの患者さんが小児期を生き延び、成人期以降も生存し社会参加の道を探ることが可能です。一方で重篤な内臓合併症がある場合の生命予後についても、率直に共有します。
- ➤指定難病185制度の活用:診断基準と重症度基準を満たす場合、医療費助成の対象となります。手続きには医師の診断書が必要です。
- ➤患者レジストリへの登録:国際的に「CSS/BAF-related disorders registry」や「CARE4ARID1B」など、患者レジストリの構築が進んでいます。長期的な自然歴データの蓄積が、将来の治療開発の基盤となります。
- ➤家族の心理的サポート:希少疾患の診断は、家族の生活と心理に大きな影響を与えます。継続的な心理的支援と情報提供が重要です。
よくある誤解と正しい理解
誤解①「親に問題があった」
CSS2の大部分は新生突然変異であり、両親の遺伝子に問題があったわけではありません。受精時または胚発生のごく初期に偶然生じた変異が原因で、誰のせいでもありません。
誤解②「ARID1A変異=CSS2と決まっている」
ARID1Aは体細胞変異としてあらゆるがんで頻発する遺伝子でもあります。生殖細胞系列変異(生まれつきの変異)と体細胞変異(がん組織だけの変異)は意味がまったく異なります。
誤解③「小指の爪が正常ならCSSではない」
分子学的に確定診断された患者さんの中にも、爪の異常が極めて軽微または存在しない非定型例が確認されています。爪の異常がないだけでCSSの可能性を排除することはできません。
誤解④「全員に同じ症状が出る」
CSS2の表現型は極めて広いスペクトラムを持ちます。体細胞モザイクの程度や、ARID1Aを補う他の経路の働きの個人差により、重症度は患者ごとに大きく異なります。
9. 臨床遺伝専門医からのメッセージ
CSS2は、特徴的な外表所見(小指の爪・粗な顔貌)から比較的疑いやすい疾患ですが、診断確定後の道のりこそが本当の意味で重要です。とくにCSS2は、他のCSSサブタイプとは違って肝芽腫リスクという独自の腫瘍感受性を伴います。これは「診断がゴールではなくスタート」であることを象徴しています。
また、近年明らかになった成人期の表現型変化——過体重・脊柱側弯・視力障害・行動異常——は、CSS2が決して「子どものうちに完結する病気」ではなく、生涯にわたる伴走型のケアを必要とする疾患であることを示しています。小児科医から内科医・整形外科医・精神科医へと、医療のバトンを切れ目なくつないでいく仕組みづくりが、これからの臨床現場に求められています。
情報がご家族の手元にあること、そして臨床遺伝専門医と継続的につながる関係を築くこと——これがCSS2のお子さんを持つご家族にとって、最も確かな道しるべになると考えています。
よくある質問(FAQ)
🏥 希少疾患の診断・遺伝カウンセリングについて
CSS2をはじめとする希少遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] Vergano SA, Deardorff MA. Coffin-Siris Syndrome. GeneReviews® [Internet]. University of Washington, Seattle. [NCBI Bookshelf NBK131811]
- [2] OMIM #614607. Coffin-Siris Syndrome 2; CSS2. Johns Hopkins University. [OMIM]
- [3] Orphanet. Coffin-Siris syndrome. ORPHA:1465. [Orphanet]
- [4] Tsurusaki Y, Okamoto N, Ohashi H, et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet. 2012;44(4):376-378. [PubMed]
- [5] Santen GW, Aten E, Vulto-van Silfhout AT, et al. Coffin-Siris syndrome and the BAF complex: genotype-phenotype study in 63 patients. Hum Mutat. 2013;34(11):1519-1528. [PubMed]
- [6] Cárcamo B, Bourlon MT, Roca-Argente L, et al. Cancer in ARID1A-Coffin-Siris syndrome: Review and report of a child with hepatoblastoma. Am J Med Genet A. 2022. [PubMed]
- [7] Schmetz A, Lüdecke HJ, Surowy H, et al. Delineation of the adult phenotype of Coffin-Siris syndrome in 35 individuals. Hum Genet. 2024. [PubMed]
- [8] Pediatric Cancer Predisposition and Surveillance. AACR Cancer Progress Report. [AACR]
- [9] Bögershausen N, Wollnik B. Mutational Landscapes and Phenotypic Spectrum of SWI/SNF-Related Intellectual Disability Disorders. Front Mol Neurosci. 2018;11:252. [Frontiers]
- [10] コフィン・シリス症候群(指定難病185). 難病情報センター. [難病情報センター]
- [11] Khella MS, Salem AM, Abdel-Rahman O, Saad AS. Systemic and ocular manifestations of a patient with mosaic ARID1A-associated Coffin-Siris syndrome. Am J Ophthalmol Case Rep. 2022. [PMC8808370]






