目次
- 1 1. スルファターゼ遺伝子群とは——硫酸基を切り離す17の酵素ファミリー
- 2 2. SUMF1とFGly修飾——17酵素を一括起動する「マスタースイッチ」
- 3 3. ヒトの17スルファターゼ遺伝子と関連疾患の全体像
- 4 4. 単一遺伝子変異が引き起こすライソゾーム蓄積症(LSD)
- 5 5. 多種スルファターゼ欠損症(MSD)——SUMF1の故障で17酵素全部が止まる
- 6 6. SULF1/SULF2——細胞外スルファターゼによるパラダイムシフト
- 7 7. がん微小環境におけるSULF1/SULF2のパラドックス
- 8 8. 遺伝子・細胞治療の最前線——「治らない病気」が変わる
- 9 9. よくある誤解
- 10 10. 臨床遺伝専門医からのメッセージ
- 11 よくある質問(FAQ)
- 12 関連記事
- 13 参考文献

📍 クイックナビゲーション
スルファターゼ(Sulfatases)は、生体内のあらゆる分子から「硫酸基」を切り離す働きを持つ17種類の酵素ファミリーです。ヒトの体ではホルモンの調節・軟骨の維持・脳の神経保護・細胞間のシグナル伝達など、極めて多彩な役割を担っており、これらの遺伝子に異常が起きると、異染性白質ジストロフィー(MLD)・ハンター症候群・モルキオA病といった重篤な遺伝病が引き起こされます。近年、AAVベクターを用いた遺伝子治療が次々と承認・臨床試験開始の段階に入り、これまで「治らない病気」とされてきた領域が大きく変わろうとしています。
Q. スルファターゼ遺伝子群とは何ですか?まず結論だけ知りたいです
A. 細胞内・細胞外で「硫酸基」を切り離す17種類の酵素群です。ステロイドホルモン・軟骨成分・脳のミエリン・成長因子のシグナルなどを精密にコントロールしており、どれか1つでも壊れると重篤な遺伝性疾患を引き起こします。SUMF1という別の遺伝子が17酵素全部を一括起動する「マスタースイッチ」になっており、ここが壊れると全部が同時に動かなくなる多種スルファターゼ欠損症(MSD)になります。
- ➤ファミリーの全体像 → ヒトには17種類のスルファターゼがあり、7グループに分類される
- ➤活性化のボトルネック → SUMF1/FGEによる翻訳後修飾(FGly形成)が必須
- ➤関連する遺伝病 → MLD・ハンター症候群・モルキオA病・MSDなど多数
- ➤パラダイムシフト → SULF1/SULF2は細胞外でシグナル伝達を制御する新しい役割
- ➤治療の最前線 → MLD遺伝子治療が承認、MSDも2026年臨床試験予定
1. スルファターゼ遺伝子群とは——硫酸基を切り離す17の酵素ファミリー
私たちの体では、ホルモン・軟骨・脳のミエリン・細胞外マトリックスといった生命を支える分子の多くに「硫酸基」と呼ばれる小さな化学グループがくっついています。この硫酸基は、分子の働きを「オン/オフ」したり、分子同士の結合を強めたり弱めたりする分子のスイッチとして機能しています。
スルファターゼは、この硫酸基を切り離す「ハサミ」の役割を持つ酵素群です。ヒトのゲノムには現在までに17種類のスルファターゼ遺伝子が同定されており、進化の過程で機能が分化しながらも、共通の祖先遺伝子から派生したと考えられています。
💡 用語解説:硫酸基(りゅうさんき)とは
「-OSO₃⁻」という化学構造を持つ小さな化学グループです。糖・脂質・タンパク質・ホルモンなど、さまざまな生体分子に「飾り」のように付加されます。硫酸基がついているかどうかで分子の電気的性質や水になじみやすさが大きく変わるため、分子の機能を切り替える「タグ」として働きます。スルファターゼはこのタグを外す酵素であり、外す時期や場所を間違えると体内で重大な障害が起きます。
どこで・何のために働いているのか
スルファターゼ酵素群は、細胞内のさまざまな場所で異なる役割を担います。
🔬 ライソゾーム内での分解
細胞内の「ゴミ処理場」であるライソゾームで、不要になった巨大な糖鎖や脂質を分解。働かないと「ライソゾーム病」になります。
⚕️ ホルモン代謝
小胞体に結合したSTS(ステロイドスルファターゼ)は、女性ホルモンや男性ホルモンの活性化に関わります。皮膚での働きが特に重要です。
🧬 細胞外シグナル制御
SULF1/SULF2は細胞表面で成長因子(Wnt・FGF・BMP・VEGF・GDNFなど)のシグナルを制御。発生・組織再生・がんに深く関わります。
🛡️ 異物代謝(肝臓)
硫酸化と脱硫酸化のバランスにより、薬物・神経伝達物質・代謝産物の不活性化と排出を制御しています。
2. SUMF1とFGly修飾——17酵素を一括起動する「マスタースイッチ」
スルファターゼ遺伝子群を理解するうえで最も重要なのは、これら17の酵素が「単独では絶対に動かない」という事実です。リボソームでアミノ酸鎖が翻訳されただけでは触媒活性を持たず、ある特殊な「翻訳後修飾」を受けて初めて働けるようになります。
💡 用語解説:翻訳後修飾(ほんやくごしゅうしょく)
タンパク質はDNAの情報をもとに合成されますが、合成後にさらに化学的な「飾り」や「加工」が加えられることがあります。これを翻訳後修飾と呼びます。リン酸化・糖鎖付加・切断などが代表例で、タンパク質に正しい働きをさせるために必須です。スルファターゼでは、活性部位の「システイン」というアミノ酸を「ホルミルグリシン(FGly)」という特殊なアミノ酸に変換する独特な修飾を受けます。
FGly修飾——スルファターゼ独自の活性化メカニズム
17すべてのスルファターゼには、活性部位の中心に特定のシステイン残基が存在します。このシステインが、SUMF1遺伝子がコードするFGE(ホルミルグリシン生成酵素)によって酸化され、Cα-ホルミルグリシン(FGly)という特殊なアミノ酸に変換されることで、初めてスルファターゼは硫酸基を切る能力を獲得します。
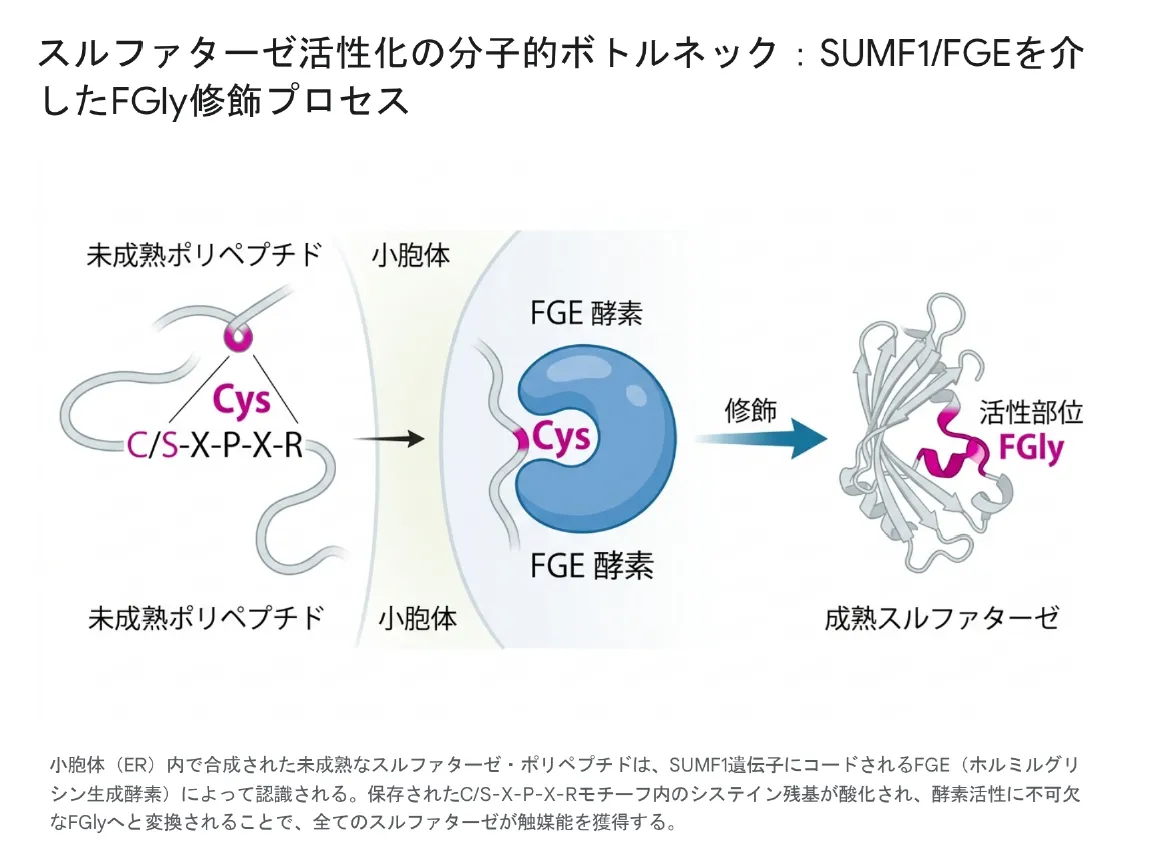
小胞体(ER)内で合成された未成熟なスルファターゼ・ポリペプチドは、SUMF1遺伝子にコードされるFGE(ホルミルグリシン生成酵素)によって認識される。保存されたC/S-X-P-X-Rモチーフ内のシステイン残基が酸化され、酵素活性に不可欠なFGlyへと変換されることで、すべてのスルファターゼが触媒能を獲得する。
🧪 FGly修飾の流れ
(C/S-X-P-X-Rモチーフ)
(SUMF1由来酵素)
(成熟スルファターゼ)
💡 用語解説:C/S-X-P-X-Rモチーフ
FGEがスルファターゼを認識するための「目印配列」です。システイン(C)またはセリン(S)から始まる、X-P-X-Rという5アミノ酸の並びを意味します。この配列は17すべてのスルファターゼで強く保存されており、進化の過程でほとんど変化していません。逆に言えば、この配列さえあればFGEに認識されて活性化される、というシンプルかつ厳密なシステムです。
なぜこのメカニズムがそれほど重要なのか
SUMF1/FGEは「17酵素全部の活性化を一手に引き受ける唯一の制御点」です。これが進化的に保存されている理由は、生体が硫酸代謝を一括管理できる効率性にあります。しかし同時に、これはSUMF1が壊れると17酵素全部が同時に機能不全に陥るという致命的な脆弱性にもなっています(後述する多種スルファターゼ欠損症)。
3. ヒトの17スルファターゼ遺伝子と関連疾患の全体像
ヒトゲノムに存在する17のスルファターゼ遺伝子は、系統発生的解析と構造類似性に基づいて7つのグループ(Group 1〜7)に分類されます。染色体上の分布にも進化の痕跡が見られ、ARSD・ARSL(旧ARSE)・ARSF・ARSH・STSはX染色体上のステリルスルファターゼ遺伝子クラスターに連続して並んでおり、過去の遺伝子重複イベントの強い証拠となっています。
📝 補足:HGNCでは18遺伝子、なぜ「17」?
HGNC(HUGO Gene Nomenclature Committee)のスルファターゼ・ファミリー(Group 410)には18遺伝子が登録されており、本記事の17に加えてARSC2(HGNC:716)が含まれます。ただしARSC2は1986年にステロイドスルファターゼ(STS)から生化学的に分離された「f型アイソザイム」として記載された歴史的概念で、現代まで分子レベルでのクローニング・機能確認がなされておらず、GeneCardsでも”Genetic Locus”(遺伝子座)扱いです。このため、Stresslerら(2022)をはじめとする最新の標準的レビューでは機能的に確立された17遺伝子を「ヒトスルファターゼ」として扱うのが慣例です。本記事もこの学術慣例に準拠しています。
17酵素のうち、ARSI・ARSJ・ARSKなど約3分の1は依然として生理的機能が完全には解明されていません。これらは「オーファン(孤児)スルファターゼ」と呼ばれ、現在も活発に研究が進められている領域です。
4. 単一遺伝子変異が引き起こすライソゾーム蓄積症(LSD)
スルファターゼの大部分は、細胞内の「ゴミ処理場」であるライソゾームに局在します。たった1つのスルファターゼ遺伝子に病的な変異が生じると、特定の硫酸化分子が分解されずにライソゾーム内に異常蓄積し、重篤なライソゾーム蓄積症(LSD)を引き起こします。
💡 用語解説:ライソゾーム蓄積症(LSD)
細胞内の小器官「ライソゾーム」で働く酵素の遺伝的欠損により、本来分解されるべき分子(糖鎖・脂質など)が細胞内に過剰に蓄積する一群の疾患です。「ムコ多糖症(MPS)」「スフィンゴリピドーシス」などが含まれ、現在約70疾患が知られています。多くは進行性で、神経系・骨格・内臓に重篤な症状を引き起こします。
代表的なスルファターゼ関連LSD
🧠 異染性白質ジストロフィー(MLD)
原因:ARSA遺伝子欠損
脳の神経を覆うミエリン鞘の主要成分「スルファチド」が分解できずに蓄積。進行性の脱髄、運動失調、痙縮、認知症を引き起こし、小児型は10歳未満で致死的に進行します。
🦴 マロトー・ラミー症候群(MPS VI)
原因:ARSB遺伝子欠損
「デルマタン硫酸」が全身に蓄積。重度の骨格変形、内臓肥大、心臓弁膜症を引き起こします。神経障害を伴わないことが多いのが特徴です。
🧒 モルキオA病(MPS IVA)
原因:GALNS遺伝子欠損
軟骨と骨の主成分「ケラタン硫酸・C6硫酸」が蓄積し、成長板を破壊。重度の骨格異形成、著しい低身長、関節弛緩、気管狭窄、心臓合併症を引き起こします。
👦 ハンター症候群(MPS II)
原因:IDS遺伝子欠損(X連鎖性)
「デルマタン硫酸・ヘパラン硫酸」が全身に蓄積。粗な顔貌、精神運動発達遅滞、肝脾腫、骨格異常を呈する。男児に多発する代表的なMPS。
🐟 X連鎖性魚鱗癬(XLI)
原因:STS遺伝子欠損
皮膚の「コレステロール硫酸」が分解できず蓄積。皮膚の異常角化(鱗屑)、角質層の剥離障害が出現。妊娠期の難産リスクを高めることでも知られます。
👂 アッシャー症候群4型
原因:ARSG遺伝子欠損
進行性の難聴と網膜色素変性が組み合わさった疾患。神経細胞死や行動障害も報告されており、聴覚と視覚の両方が侵されます。
💡 用語解説:グリコサミノグリカン(GAG)
長い糖の鎖のことで、軟骨・皮膚・血管壁などの「クッション」や「水分保持材」として体中で使われています。デルマタン硫酸・ヘパラン硫酸・ケラタン硫酸などが代表例。スルファターゼはGAGから硫酸基を切り離す働きを持ちますが、これができないとGAGがライソゾームに溜まり続け、ムコ多糖症(MPS)と総称される疾患群を引き起こします。

5. 多種スルファターゼ欠損症(MSD)——SUMF1の故障で17酵素全部が止まる
個々のスルファターゼ遺伝子変異が「単一の疾患」を引き起こすのに対し、マスタースイッチであるSUMF1の病的変異は、これらすべての疾患が同時多発的に襲いかかるという壊滅的な結果をもたらします。これが多種スルファターゼ欠損症(Multiple Sulfatase Deficiency: MSD)です。
⚠️ MSDの基本データ
- 発症率:50万人に1人未満(超希少疾患)
- 原因:SUMF1遺伝子変異によるFGE機能不全
- 結果:17スルファターゼ全部が活性を持たないまま組織に到達
- 承認された疾患修飾療法:現時点でなし
なぜ多臓器が同時崩壊するのか
MSDの患者体内では、FGEの機能不全により17種類すべてのスルファターゼが小胞体を通過した段階で触媒活性を獲得できません。これにより以下が同時に進行します。
- ① ムコ多糖症(MPS)的所見:全身細胞へのGAG蓄積、粗な顔貌、骨格異形成、肝脾腫
- ② MLD的所見:神経系へのスルファチド蓄積、進行性の脱髄、てんかん、運動退行
- ③ X連鎖性魚鱗癬的所見:皮膚へのステロイド硫酸蓄積、皮膚異常
3つの臨床サブタイプ
新生児型(最重症型)
出生前から病態が進行。子宮内発育遅延・出生直後からの重篤な呼吸窮迫を示します。残存酵素活性が極めて低い症例。
乳児型(最頻度型)
幼児期に定型的な退行を示します。歩行障害・痙縮・運動失調・自閉症的特徴・てんかんに加え、骨格異形成・肝脾腫・水頭症・失明・難聴が累積していきます。
若年型
進行がやや緩やかな稀なサブタイプ。確定診断の難しさから発見バイアスがかかっている可能性も指摘されています。
最近の研究では、MSDの表現型(重度・減弱型)が特定のSUMF1遺伝子型と、新たなバイオマーカーである「グリコサミノグリカン非還元末端(GAG-NREs)」のプロファイルに強く相関することが明らかになりつつあり、臨床試験のエンドポイント設定の鍵として注目されています。
6. SULF1/SULF2——細胞外スルファターゼによるパラダイムシフト
従来、スルファターゼは「ライソゾーム内での不要物の分解酵素」として認識されてきました。しかし、SULF1とSULF2の発見は、このファミリーの機能を「細胞間シグナル伝達の精密なエピジェネティック制御」へとパラダイムシフトさせました。
💡 用語解説:ヘパラン硫酸プロテオグリカン(HSPG)
細胞表面や細胞外マトリックスに存在する、長い「ヘパラン硫酸」と呼ばれる糖鎖がタンパク質に結合した分子です。Wnt・FGF・BMP・VEGF・GDNFなどの成長因子と結合し、これらのシグナル分子の「貯蔵庫」「共受容体」「拡散コントローラ」として機能します。SULF1/SULF2はこのHSPGの硫酸基を選択的に外すことで、シグナル伝達の強さや方向性を細かく制御します。
「ピンポイント編集」というユニークな働き
SULF1とSULF2は、HSPG糖鎖の内部「S-ドメイン」に存在する特定の二糖単位のC-6位から硫酸基を選択的に除去します。このピンポイントな構造改変(エディティング)により、HSPGとシグナル分子との相互作用が物理的に変化し、結合部位が変容します。
結果として、成長因子がHSPGから細胞外空間へと放出されて下流のシグナル経路を活性化したり、逆に共受容体との三者複合体形成が阻害されてシグナルがダウンレギュレーションされたりと、極めてダイナミックかつ文脈依存的な調節が行われます。
SULF欠損が引き起こす生体異常
食道神経支配の破綻
SULF1/SULF2のダブルノックアウトマウスでは、GDNFがHSPGに捕捉されたまま神経細胞に届かず、食道の腸管グリア形成不全と収縮機能不全という致命的異常が発生します。
軟骨恒常性の崩壊
SULF欠損マウスではBMP/FGFバランスが崩壊し、軟骨基質因子の発現低下と分解酵素の上昇により、変形性関節症に似た病理が早期から進行します。
Wnt経路のスイッチング
SULF1はカノニカルWnt経路を阻害する一方、非カノニカルWnt経路を増強。シグナルを「振り分けるスイッチャー」として機能します。
7. がん微小環境におけるSULF1/SULF2のパラドックス
発生や恒常性維持に不可欠なSULFのシグナル制御機構は、悪性腫瘍においてハイジャックされ、がん細胞の増殖・浸潤・血管新生を促進する強力なオンコジーン(発がん遺伝子)としての顔を持ちます。
がん細胞と線維芽細胞の「分業体制」
最新の単一細胞RNAシーケンス解析と患者由来異種移植モデルによる検証により、SULF1とSULF2が由来する細胞ソースが空間的に完全に分離していることが証明されました。
SULF1の供給源
腫瘍周囲の「がん関連線維芽細胞(CAF)」から選択的に供給。腫瘍細胞ではほとんど発現しません。疾患の初期の悪性化と生存率低下を駆動します。
SULF2の供給源
主に「腫瘍細胞自身」から生産・分泌されます。後期の進行を牽引し、PDGFR・VEGFなど受容体チロシンキナーゼ(RTK)経路を活性化します。
特定のがん種における過剰発現
📊 特定がん種におけるSULF1/SULF2の発現上昇(Log2 Fold Change)
頭頸部扁平上皮癌・膠芽腫など多数のがん種で過剰発現が確認されており、患者の予後不良と強く相関します
一方で、嫌色素性腎細胞癌や甲状腺癌ではSULF1が、前立腺癌ではSULF2が有意にダウンレギュレーションされており、一部の乳がん・多発性骨髄腫モデルではSULF1/SULF2が強力な腫瘍増殖阻害剤として機能する報告も存在します。これらの機能的二面性は、SULFの作用が標的組織のHSPG組成や依存するシグナル経路(文脈)に強く依存することを示しています。
8. 遺伝子・細胞治療の最前線——「治らない病気」が変わる
スルファターゼ関連疾患の治療は、過去数十年間、外部から不足した酵素を定期的に点滴投与する「酵素補充療法(ERT)」が主流でした。しかし、ERTには重大な限界があります。
⚠️ ERTの限界
- 血液脳関門(BBB)を越えられないため、MLDやMSDの中枢神経変性に無力
- 定期点滴の継続投与が生涯必要
- MSDのように17酵素が同時不全の疾患には個別補充は不可能
クロスコレクション原理——遺伝子治療を可能にしたブレイクスルー
💡 用語解説:クロスコレクション(cross-correction)
遺伝子改変によって正常な酵素を過剰発現・分泌する細胞を体内に定着させると、そこから分泌された活性酵素がマンノース-6-リン酸(M6P)受容体を介して隣接する病的な細胞に自律的に取り込まれ、その機能を補正する原理です。1つの「治療済み細胞」が周囲の何百もの「病気の細胞」を救えるため、遺伝子治療の実用化を可能にした基盤理論です。
承認済み:MLDに対するEx vivo遺伝子治療(Libmeldy/Lenmeldy)
ARSA欠損症であるMLDに対し、患者自身の造血幹細胞(HSC)を体外に取り出し、レンチウイルスベクターで正常なARSA遺伝子を挿入してから戻す治療が確立されました。改変HSCはマクロファージやミクログリア系統に分化して血液脳関門を越え、中枢神経系で持続的に酵素を分泌します。
🎉 歴史的快挙
- 2020年:欧州EMAでLibmeldy®が正式承認
- 2024年初頭:米国FDAでLenmeldy™が正式承認
- 条件:発症前または初期症状の段階で投与された場合、治癒的効果
MSDに対する革新的アプローチ——BGTC枠組み
SUMF1変異によるMSDは長らく治療法開発の蚊帳の外でしたが、フィラデルフィア小児病院(CHOP)等のチームがMSDモデルマウスとMSD患者由来幹細胞を用いたEx vivoレンチウイルスHSC遺伝子治療の前臨床試験で有望な結果を示しています。AAV9-SUMF1ベクターをマウスの脳室および全身に投与した研究では、脳を含む全身でスルファターゼのグローバルな活性化が確認され、GAGのほぼ完全なクリアランスと行動表現型の劇的改善が報告されました。
💡 用語解説:BGTC(Bespoke Gene Therapy Consortium)
2021年10月、米国NIH・FDA・製薬企業・患者団体が連携して設立された官民パートナーシップ。1億ドル以上の資金のもと、AAV遺伝子治療の規制経路標準化を推進しています。初期臨床ポートフォリオ8疾患のうち2つに、MSDとモルキオ症候群が選定されました。CHOPが主導するMSD自然歴調査が進行中で、早ければ2026年初頭にもMSDに対するAAV遺伝子治療の臨床試験開設が予定されています。
モルキオA病(MPS IVA)についても、ラットモデルにおけるAAV9-Galnsベクター投与研究で、1年という長期にわたり骨・軟骨・気管・心臓を含む末梢組織全体での酵素活性上昇とKSレベルの正常化が達成され、深刻な骨格異形成を予防する強力な治療効果が実証されました。
9. よくある誤解
誤解①「スルファターゼは1種類の酵素」
スルファターゼはヒトに17種類存在する酵素ファミリーです。それぞれが異なる基質(脂質・糖鎖・ホルモンなど)を分解しており、壊れる遺伝子によって全く違う病気になります。
誤解②「酵素補充療法ですべて治る」
ERTは血液脳関門を越えられないため、中枢神経症状には無力です。MLDやMSDの脳症状にはAAVや造血幹細胞ベースの遺伝子治療が必要になります。
誤解③「成長して治る発達遅滞」
スルファターゼ関連疾患の多くは「発達退行(獲得した能力を失う)」を示します。「いずれ追いつく」発達遅滞ではなく、早期介入が予後を左右する進行性疾患です。
誤解④「親が健康なら遺伝病ではない」
スルファターゼ関連疾患の多くは常染色体潜性(劣性)遺伝。両親が健康でも、保因者同士の組み合わせで4分の1の確率で発症します。キャリアスクリーニングが家族計画の鍵です。
10. 臨床遺伝専門医からのメッセージ
スルファターゼという酵素群は、生命現象における「硫酸基をどう動かすか」という極めて精密な制御を担っています。たった1つの酵素が止まるだけで全身が崩壊する疾患があり、17全部が同時に止まる疾患があり、本来は静かに働く酵素ががん細胞に乗っ取られて増殖を加速する事例もあります。
同時に、SUMF1を1つ修復すれば17酵素全部が同時に活性化されるという構造的特徴は、これらの疾患を「遺伝子治療で一度にリブートできる」可能性を提供してくれています。MLD遺伝子治療の承認、MSDのBGTC臨床試験開始(2026年予定)、モルキオA病のAAV治療進展——これらは数年前まで想像もできなかった現実です。
私たち臨床遺伝専門医にとって最大の使命は、「治療可能な時間軸」のうちに正確な診断にたどり着くことです。原因不明の発達退行・骨格異形成・進行性運動障害を見たとき、スルファターゼ関連疾患を鑑別の引き出しから取り出してください。NGSパネル検査・全エクソーム解析が、その答えを最速で出してくれます。
よくある質問(FAQ)
🏥 スルファターゼ関連疾患・遺伝子検査について
MLD・ハンター症候群・モルキオA病・MSDなどの希少遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] Diez-Roux G, Ballabio A. Sulfatases and human disease. Annu Rev Genomics Hum Genet. 2005;6:355-379. [PubMed]
- [2] Sardiello M, et al. Sulfatases and sulfatase modifying factors: an exclusive and promiscuous relationship. Hum Mol Genet. 2005;14(21):3203-3217. [Oxford Academic]
- [3] Stressler T, Eisele T. Mammalian Sulfatases: Biochemistry, Disease Manifestation, and Therapy. Int J Mol Sci. 2022. [PMC9330403]
- [4] Adang LA, et al. Multiple Sulfatase Deficiency. GeneReviews®. 2020. [GeneReviews/NCBI]
- [5] Schlotawa L, et al. Long-term disease course of two patients with multiple sulfatase deficiency differs from metachromatic leukodystrophy. JIMD Rep. 2021. [PMC7932862]
- [6] Otto NM, et al. Sulfation Pathways During Neurodevelopment. Front Mol Neurosci. 2022. [PMC9047184]
- [7] Helmholz H, et al. Sulfatases: Critical Enzymes for Algal Polysaccharide Processing. Front Plant Sci. 2022. [Frontiers]
- [8] Otsuki T, et al. Extracellular sulfatases support cartilage homeostasis by regulating BMP and FGF signaling pathways. PNAS. 2010;107(22):10202-10207. [PNAS]
- [9] Ai X, et al. SULF1 and SULF2 regulate heparan sulfate-mediated GDNF signaling for esophageal innervation. Development. 2007;134(18):3327-3338. [Development]
- [10] Flowers SA, et al. Extracellular Heparan 6-O-Endosulfatases SULF1 and SULF2 in Head and Neck Squamous Cell Carcinoma and Other Malignancies. Cancers. 2022. [PMC9688903]
- [11] Phillips JJ, et al. Heparan sulfate sulfatase SULF2 regulates PDGFRα signaling and growth in human and mouse malignant glioma. J Clin Invest. 2012;122(3):911-922. [JCI]
- [12] Penati R, et al. Metachromatic Leukodystrophy: New Therapy Advancements and Emerging Research Directions. Int J Mol Sci. 2025. [PMC12205745]
- [13] Bertolin J, et al. Treatment of skeletal and non-skeletal alterations of Mucopolysaccharidosis type IVA by AAV-mediated gene therapy. Nat Commun. 2021. [PMC8429698]
- [14] Spampanato C, et al. Entering the playing field: Therapy for multiple sulfatase deficiency. Mol Ther. 2024. [PMC11573562]
- [15] NCATS-NIH. Bespoke Gene Therapy Consortium (BGTC). [NCATS]
- [16] United MSD Foundation. Bespoke Gene Therapy Consortium award. [curemsd.org]






