目次

ギオン・アルメイダ型下顎顔面異骨症(MFDM)は、EFTUD2という遺伝子の片方が働きを失うことによって起こる、100万人に1人未満という非常にまれな先天性の症候群です。下あごや頬の骨の発達が弱いこと、生まれたあとに進む小頭症、特徴的な耳の形、伝音性の難聴などが組み合わさって現れます。一方で症状の重さは人によって大きく異なり、命に関わる合併症を伴う方から、診断が難しいほど軽い方まで幅広く存在します。
Q. ギオン・アルメイダ型下顎顔面異骨症とはどんな病気ですか?まず結論だけ知りたいです
A. 細胞の中でRNAを正しく編集する「スプライソソーム」という装置の部品(EFTUD2)が半分しか働かなくなることで、顔の骨や脳の発達がうまく進まなくなる、まれな先天性の症候群です。下あご・頬骨の低形成、進行性の小頭症、耳の形の異常と伝音性難聴、食道閉鎖や心疾患などの全身症状を伴うことがあります。症状の重さには大きな個人差があります。
- ➤疾患の定義 → OMIM 610536、Orphanet ORPHA:79113、有病率は100万人に1人未満
- ➤発症のしくみ → スプライソソームの部品EFTUD2のハプロ不全と、神経堤細胞のアポトーシス
- ➤主な症状 → 小頭症(約90%)・難聴(70〜80%)・食道閉鎖や心疾患(約30%)
- ➤鑑別診断 → トリーチャー・コリンズ症候群・ネイジャー症候群との違いを詳しく解説
- ➤診断・管理 → EFTUD2遺伝子解析・全エクソーム解析と多職種チーム医療
1. ギオン・アルメイダ型下顎顔面異骨症とは:定義と歴史
ギオン・アルメイダ型下顎顔面異骨症(Mandibulofacial Dysostosis, Guion-Almeida type/略してMFDGA、OMIM 610536)は、顔まわりの骨が十分に育たない「下顎顔面異骨症」に、進行性の小頭症と発達の遅れが加わることを最大の特徴とする、まれな先天性の症候群です。医学の現場では「小頭症を伴う下顎顔面異骨症(Mandibulofacial dysostosis with microcephaly:MFDM)」という名前でも広く呼ばれており、この記事でも以降は「MFDM」と表記します。
2023年に改訂された国際的な骨系統疾患の分類では、トリーチャー・コリンズ症候群やネイジャー症候群などと同じ「頭蓋顔面異骨症」のグループに位置づけられています。つまりMFDMは、顔の骨の形成不全をきたす疾患群の一員でありながら、脳や全身にも影響が及ぶ点で、それらと一線を画す存在だといえます。
💡 用語解説:下顎顔面異骨症(かがくがんめんいこつしょう)
「異骨症」とは骨の作られ方そのものに異常がある状態のこと。「下顎顔面」は、下あご・頬骨・耳など、お顔の中〜下半分の骨を指します。これらの骨は赤ちゃんがお腹の中にいるごく初期に、鰓弓(さいきゅう)と呼ばれる構造から作られます。その発達がうまくいかないために、下あごが小さい・頬骨が低い・耳の形が違う、といった特徴が現れます。
この病気が独立した疾患として最初に報告されたのは2000年のことで、ブラジルの臨床遺伝医ギオン・アルメイダ(Guion-Almeida)らが、成長の遅れ・知的障害・小頭症・口蓋裂・耳の前の皮膚のでっぱり(副耳)などを併せ持つ兄弟例を詳しく記載したのが出発点です。その後の追加報告を経て臨床像が整理され、2012年にLinesらが全エクソーム解析という手法を用いて、原因が第17番染色体(17q21.31)にあるEFTUD2遺伝子の変化であることを突き止めました。これにより、この病気が常染色体顕性(優性)遺伝の形をとることが確定しました。
💡 用語解説:常染色体顕性(優性)遺伝とは
「常染色体」は性別を決めるX・Y以外の染色体のこと。「顕性(優性)」とは、ペアになっている2本の遺伝子のうちどちらか片方に変化があるだけで症状が現れる、という意味です。MFDMでは、変化したEFTUD2を1つ持つだけで発症します。ただし後で述べるように、MFDMの多くはご両親には変化がなく、お子さんで初めて生じた新生突然変異(de novo変異)で起こります。
これまでに世界で130例以上の確定診断例が報告されており、その数は年々増えています。注意したいのは、MFDMが他の病気と間違われやすいことです。原因のはっきりしない発達の遅れとして見過ごされたり、見た目が似ているトリーチャー・コリンズ症候群の非典型例と誤って判断されたりすることがあり、実際の頻度は「100万人に1人未満」という推定よりも高い可能性が指摘されています。
2. 原因遺伝子EFTUD2と発症のしくみ
MFDMの根本的な原因は、EFTUD2遺伝子の機能喪失型の変化にともなうハプロ不全です。EFTUD2は28個のエクソンからなり、進化の過程でほとんど変わらずに保たれてきた、972個のアミノ酸でできた116kDaのタンパク質(GTPアーゼ)の設計図です。このタンパク質は、細胞の核の中で働く「U5 snRNP」という装置の、欠かせない部品として働いています。
スプライソソームという「mRNAの編集装置」
遺伝子からタンパク質が作られるとき、いったんDNAの情報がmRNA(メッセンジャーRNA)として写し取られます。ところが写したばかりのmRNAには、最終的に必要な部分(エクソン)と不要な部分(イントロン)が混ざっています。この不要な部分を正確に切り取ってつなぎ合わせる作業をスプライシングといい、それを担う巨大な装置がスプライソソームです。EFTUD2が作る部品は、このスプライソソームが正しく動くための、いわば「中心の歯車」です。
💡 用語解説:スプライシングとスプライソソーム
スプライシングは、mRNAの「下書き」から不要な部分を切り取り、必要な部分だけをつなぎ直して「完成原稿」にする編集作業です。その編集を行う分子の複合体がスプライソソーム。EFTUD2はその主要部品の一つで、編集の途中での組み替えや、作業後の片づけ・再利用といった、とても動的な工程に欠かせません。スプライシングの基礎はこちらの用語ページでも解説しています。
💡 用語解説:ハプロ不全(はぷろふぜん)
私たちは同じ遺伝子をふつう2本(ペア)持っています。多くの遺伝子は片方が壊れても、もう片方が頑張ってくれるので問題は起きません。ところがEFTUD2のように、2本そろってこそ十分な量が確保できる遺伝子では、片方が働きを失うと量が足りなくなり、症状が出てしまいます。これがハプロ不全です。くわしくはハプロ不全の用語ページもご参照ください。
なぜ「顔」と「脳」だけに強く出るのか:神経堤細胞のパラドックス
スプライシングは全身のあらゆる細胞でいつも行われている、生命に必須の作業です。それなのに、EFTUD2のハプロ不全が顔の骨格と脳神経系という、ごく限られた場所にだけ強い影響を出すのはなぜか——これは長く「スプライソソーム病のパラドックス」と呼ばれてきた大きな謎でした。
💡 用語解説:神経堤細胞(しんけいていさいぼう)
胎児のごく初期にだけ現れる、特別な「移動する細胞集団」です。体のあちこちへ旅をしながら、軟骨・骨・末梢神経などさまざまな細胞へと姿を変えます。下あご・頬骨・中耳の小さな骨などは、この神経堤細胞からつくられます。移動と変身の最中の神経堤細胞は、遺伝子の読み替えを猛烈な勢いで行う必要があるため、スプライシング機構への「依存度」が他の細胞よりずっと高いのです。
ゼブラフィッシュやカエルを使った研究から、このしくみが分子レベルで見えてきました。EFTUD2が半分しか働かずスプライシングの効率が落ちると、神経堤細胞の中に不完全なmRNAが大量にたまります。本来は異常なmRNAを掃除する「NMD」というしくみが働くはずですが、効率の低下でこの防御も追いつかなくなります。その結果、細胞に強いストレスがかかりp53という経路が過剰に活性化。移動中・変身中の神経堤細胞が特異的にアポトーシス(プログラムされた細胞死)を起こしてしまいます。この「顔と脳のもとになる細胞の大量死」こそが、下顎顔面異骨症や小頭症の直接の引き金だと考えられています。
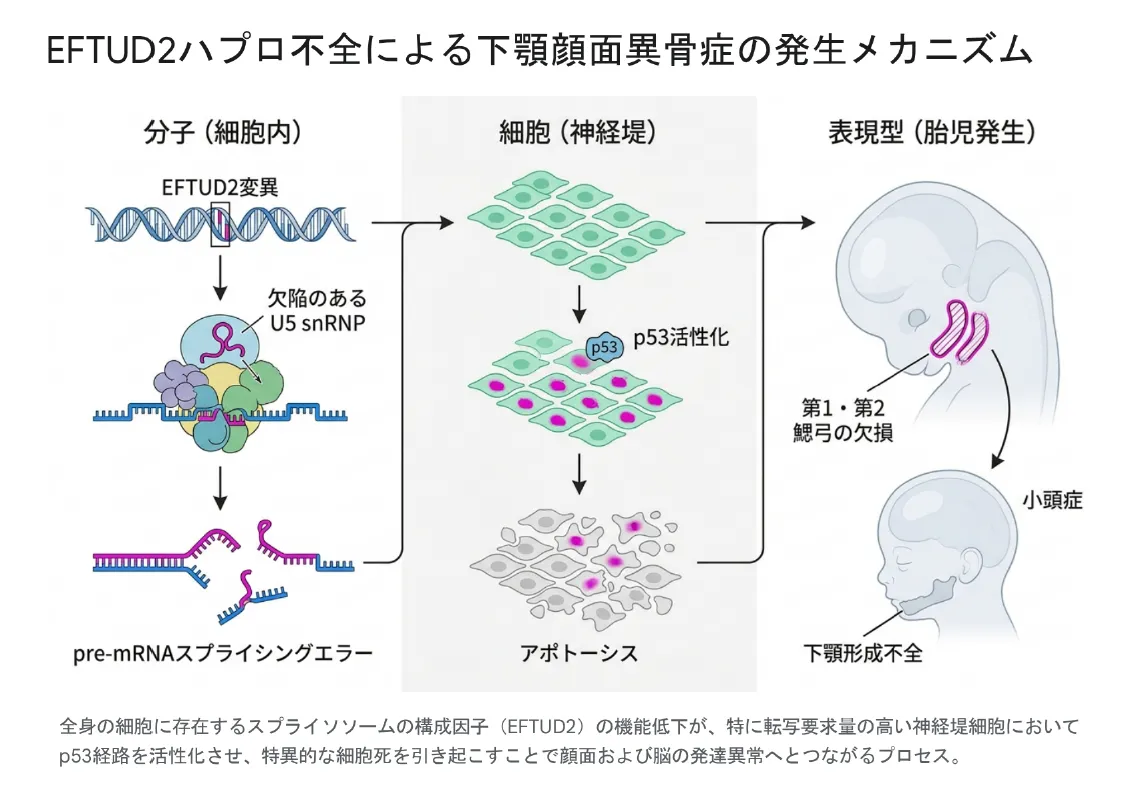
全身の細胞にあるスプライソソームの部品(EFTUD2)の働きが半分に低下すると、特に読み替えの需要が大きい神経堤細胞でp53経路が活性化し、特異的な細胞死が起こることで、顔と脳の発達異常につながる。
変化のタイプ:「タンパク質が作られない」型が大多数
これまでに見つかっているEFTUD2の病的な変化は160種類以上にのぼり、1か所だけの変化から、エクソン単位・遺伝子まるごとの小さな欠失まで多岐にわたります。データベースの集計では、フレームシフト変異が約28%、スプライス部位の変異が約26%、ナンセンス変異が約14%、大きな欠失が約10%を占める一方、アミノ酸が1つ入れ替わるミスセンス変異は約18〜19%と比較的少ないのが特徴です。つまり「タンパク質がまったく作られない・短くて働かないものになる」タイプが圧倒的に多く、これは『量が足りなくなるハプロ不全』が病気の主役であるという考えを強く裏づけています。
💡 用語解説:変化(変異)のいろいろな種類
ミスセンス変異=アミノ酸が1つ別の種類に置き換わる変化(解説)。
ナンセンス変異=途中で「ここで終わり」の合図が入り、短いタンパク質しかできない変化(解説)。
フレームシフト変異=塩基の足し引きで「読む枠」がずれ、それ以降が意味をなさなくなる変化(解説)。

3. 主な症状と全身の合併症
MFDMの症状は全身の複数の臓器におよびます。報告されている患者さんのうち約59%が男性で、やや男性に多い傾向があります。まず主な症状とおおよその出現頻度を見てから、システムごとに詳しく解説します。
MFDMの主な症状と推定される出現頻度
顔の骨格や小頭症はほぼ必発である一方、食道閉鎖や心疾患などの命に関わる合併症も約3割の方に起こります。
システム別にみた主な症状
🧠 脳・神経・発達
- 小頭症:約90%(生後に進むタイプも多い)
- 知的・運動発達の遅れ、とくに言葉の遅れ
- てんかん:約26%
- 自閉スペクトラム症・睡眠障害の併発も報告
👃 顔の骨格・気道
- 下あごの低形成(小顎症)・頬骨と中顔面の低形成
- 後鼻孔閉鎖:新生児の呼吸困難の原因
- 口蓋裂の合併も多い
🦻 耳・聴覚
- 耳介の異形成・副耳・外耳道の狭窄や閉鎖
- 難聴:70〜80%以上(伝音性が主体)
- 耳たぶの後下縁が「直角状」という特徴も
🫀 消化器・心臓・骨格
- 食道閉鎖・気管食道瘻:27〜35%
- 先天性心疾患:30〜35%
- 低身長・脊椎の異常・母指の異常
💡 用語解説:後鼻孔閉鎖(こうびこうへいさ)
鼻のいちばん奥(のどへの出口)が骨や膜でふさがっている状態です。新生児は基本的に鼻でしか呼吸できないため、両側がふさがっていると生まれた直後から命に関わる呼吸困難を起こし、緊急の気道確保が必要になります。MFDMでは小さな下あご(小顎症)による舌の落ち込みも重なり、新生児期の呼吸管理がとても重要です。
💡 用語解説:食道閉鎖・気管食道瘻(しょくどうへいさ・きかんしょくどうろう)
食道の途中が行き止まりになり、本来つながっていない気管と異常につながってしまう状態です。最も多い「C型」では、飲み込んだ母乳や唾液が肺に流れ込み、重い誤嚥性肺炎を起こします。出生前には羊水が多い(羊水過多)・超音波で胃の袋が見えにくい、といったサインで疑われることがあります。新生児期の外科手術が必要な、命に直結する合併症です。
症状の重さには大きな個人差がある
MFDMは、同じEFTUD2の変化があっても人によって症状の出方や重さが大きく違うことが知られています(表現度の多様性)。命に関わる多臓器の奇形を伴う重い例から、診断が難しいほど軽い例まで連続しています。さらに最近のコホート研究では、フレームシフト変異を持つ方でミスセンス変異の方より先天性心疾患の合併率が高いという、遺伝子型と症状の関連(遺伝子型-表現型相関)も指摘されています。
4. 鑑別診断:似た病気との見分け方
MFDMの診断でいちばん大切なのは、顔まわりの所見がよく似た他の病気と正確に見分けることです。見た目が似ていても、根本のしくみや合併症のプロファイル、そして予後が大きく異なるため、その後の管理方針を左右します。
トリーチャー・コリンズ症候群(TCS)との違い
下あご・頬骨の低形成、小耳症、外側に下がる目もと、伝音性難聴など、顔まわりはMFDMと非常によく似ています。ただし原因はリボソームを作るための遺伝子(TCOF1、POLR1C、POLR1Dなど)で、スプライシングとは無関係です。
見分けの決め手:TCSでは通常、知能は正常で小頭症を伴いません。食道閉鎖や心疾患、母指の異常もまれです。知的障害と小頭症の有無が両者を分ける鍵になります。
ネイジャー症候群との違い
重い下顎顔面異骨症に加えて「四肢の異常」を伴う病気で、多くはSF3B4遺伝子が原因です。SF3B4もEFTUD2と同じくスプライソソーム(U2 snRNP)の部品で、両者は同じ「スプライソソーム病」の仲間として共通の病態を持ちます。
見分けの決め手:ネイジャー症候群では橈骨(前腕の骨)の低形成や親指の欠損など、強い上肢の異常が目立ち、ふつう小頭症や重い知的障害は伴いません。MFDMの母指異常はそこまで重くありません。
| 特徴 | ギオン・アルメイダ型(MFDM) | トリーチャー・コリンズ(TCS) | ネイジャー症候群 |
|---|---|---|---|
| 主な原因遺伝子 | EFTUD2(17q21.31) | TCOF1、POLR1C/D など | SF3B4 |
| しくみ | スプライソソーム機能不全(U5) | リボソーム生合成の異常 | スプライソソーム機能不全(U2) |
| 小頭症 | ほぼ必発(生後進行性が多い) | 正常 | 正常 |
| 知的・神経発達 | 軽度〜重度の知的障害・言語遅延 | 正常範囲 | 正常範囲 |
| 手・四肢の異常 | 軽度の母指異常(約35%)・合指症 | ほぼ見られない | 重度の橈骨・母指欠損(必発) |
| 食道閉鎖/気管食道瘻 | 27〜35%で合併 | 極めて稀 | 稀(最重症型を除く) |
5. 診断の進め方と遺伝子検査
診断は、まず詳しい診察から始まります。下顎顔面異骨症(頬骨・上あごの低形成)があり、そこに「進行性の小頭症」「軽度〜重度の知的障害」「特徴的な耳の所見(直角状の耳たぶ・副耳)」「伝音性難聴」のいずれかが加わるとき、MFDMを強く疑います。最終的な確定診断はEFTUD2遺伝子のヘテロ接合性の病的変化を見つけることで行います。
出生後の確定診断
お子さんが生まれた後の確定診断は、血液などを用いた分子遺伝学的検査で行います。症状の典型度に応じて、次のような段階的・網羅的なアプローチが選ばれます。
- ➤EFTUD2の遺伝子標的検査:典型的な症状がそろう場合、まずEFTUD2の全コード領域とスプライス部位を解析します。これだけで病的変化の約93%をカバーできます。見つからなければ、エクソン単位や遺伝子全体の大きな欠失・重複を調べる解析(MLPAなど)で残りの約7%を捉えます。
- ➤マルチ遺伝子パネル検査:トリーチャー・コリンズ症候群など似た病気との見分けを同時に行うため、関連する複数の遺伝子をまとめて調べる方法。EFTUD2はこうしたパネルにも含まれています。
- ➤全エクソーム/全ゲノム解析(WES/WGS):症状が非典型的で、発達の遅れや小頭症だけが目立ち顔の所見が軽い場合に有効です。曖昧な症例でも20〜30%で原因が判明し、誤診からの再診断につながります。
💡 用語解説:全エクソーム解析(WES)
遺伝子のうち、タンパク質の設計図になっている部分(エクソン)をまとめて読む検査です。エクソンはゲノム全体の1〜2%にすぎませんが、病気の原因の大部分がここに集中しています。とくにご両親も一緒に3人で解析する「トリオ解析」は、お子さんで初めて生じた新生突然変異を効率よく見つけられるため、MFDMのように新生突然変異が多い病気で力を発揮します。
出生前の診断について
出生前の超音波検査では、食道閉鎖に関連する羊水過多や胃の袋が見えにくい所見、強い小顎症、脳の形態異常や発達の遅れなどが警告サインになることがあります。ただしこれらは特異性が低く、超音波だけで出生前にMFDMを確定診断することは極めて困難です。
ご家族の中にすでにEFTUD2の病的変化が分かっている場合に限り、絨毛検査・羊水検査を用いた分子遺伝学的な出生前診断や、体外受精における着床前遺伝学的検査(PGT)が選択肢になります。なお、当院のNIPTのうちインペリアルプランはEFTUD2を含む多数の単一遺伝子をスクリーニングの対象としています。検査を受けるかどうか、どこまで調べるかは、ご家族の価値観に沿ってお決めいただく事柄です。
6. 治療と長期的な管理
現在のところ、変化したEFTUD2そのものを治す根本的な治療法(遺伝子治療など)はありません。そのため、新生児科・形成外科・口腔外科・耳鼻咽喉科・小児神経科・循環器科・リハビリテーション科などが連携する多職種チーム医療で、成長の段階に合わせたきめ細かな対症療法とケアを続けていくことが柱になります。
新生児期:命を守るための最優先対応
両側の後鼻孔閉鎖や強い小顎症があると、生まれた直後から重い上気道の閉塞を起こす危険があります。経鼻エアウェイの挿入、気管内挿管、ときに一時的な気管切開で気道を確保します。小さな下あごには、骨を少しずつ前に伸ばす「下顎骨延長術」が行われることもあります。また食道閉鎖・気管食道瘻があれば、誤嚥による肺の損傷を防ぐため、新生児期に外科手術が必要です。摂食・嚥下の障害が続く場合は経鼻胃管栄養や胃瘻も検討し、言語聴覚士による嚥下リハビリを並行します。
学童期以降:機能の再建と発達支援
形成・口腔外科
口蓋裂は通常1歳〜1歳半ごろに閉鎖。成長に合わせて頬骨・下あごの骨移植やかみ合わせの矯正、耳の形に対する肋軟骨を用いた再建やエピテーゼ(人工の耳)も選択肢になります。
聴覚の管理
伝音性難聴は言葉の発達を妨げる大きな要因。外耳道閉鎖などで一般的な補聴器が使いにくい場合は、骨固定型補聴器(BAHA)・軟骨伝導補聴器・重度では人工内耳の早期導入が勧められます。
発達への包括的支援
理学療法・作業療法・言語聴覚療法を組み合わせた早期介入を乳幼児期から開始。言葉が出にくいお子さんには、絵カードや音声出力機器などの代替コミュニケーション(AAC)が大きな助けになります。
解剖学的な気道の狭さから、小児期以降も閉塞性睡眠時無呼吸のリスクが続くため、睡眠検査などの定期評価が必要です。先天性心疾患が隠れていることもあり、診断時には心エコーでの確認を行います。
7. 遺伝カウンセリングと再発について
MFDMでは遺伝カウンセリングがとても重要な役割を果たします。発端者(最初に診断されたお子さん)の75%以上が新生突然変異(de novo変異)で発症するため、健康なご両親から次のお子さんに同じ病気が再び起こる確率(再発率)は、一般の方とほとんど変わらないと説明されます。
💡 用語解説:生殖細胞系列モザイク
ご両親の血液などの体の細胞には変化がなくても、精子や卵子をつくる細胞の一部にだけ同じ変化が潜んでいる状態です。MFDMでは約6%でこの可能性が確認されています。「ご両親の検査が陰性だから次子は絶対大丈夫」とは言い切れない、ということを意味する大切なポイントで、慎重なカウンセリングが求められます。
一方、診断を受けたご本人が将来お子さんを持つ場合は、常染色体顕性(優性)遺伝の原則に従い、お子さんへ受け継がれる確率は理論上50%です。遺伝子の変化がすでに分かっている家系では、次の妊娠で超音波による形態モニタリングに加えて、ご希望に応じて絨毛検査・羊水検査による出生前診断やPGTといった選択肢も提供できます。
8. よくある誤解
誤解①「顔が似ているからトリーチャー・コリンズだ」
顔まわりはとてもよく似ますが、小頭症や知的障害があればMFDMを疑うべきです。原因遺伝子も病態も異なり、合併症のリスクも違います。
誤解②「発達がゆっくりなだけ」
発達の遅れの背景に、後鼻孔閉鎖・食道閉鎖・心疾患など命に関わる合併症が隠れていることがあります。一つの症候群として全身を評価することが大切です。
誤解③「両親が健康だから遺伝ではない」
MFDMの多くは新生突然変異で、ご両親には同じ変化がありません。「遺伝ではないはず」という思い込みが、診断を遅らせることがあります。
誤解④「予後は遺伝子で決まる」
予後は遺伝子の変化そのものより、食道閉鎖・心疾患・新生児期の気道障害などの合併症の重さに大きく左右されます。早期の適切な介入が将来を支えます。
9. 臨床遺伝専門医からのメッセージ
MFDMは、全身のあらゆる細胞で働くはずの基本的なRNAの編集機構が少し低下するだけで、なぜ顔と脳という特定の場所にだけ大きな影響が出るのか——という長年の謎を抱えてきました。その答えが、神経堤細胞という特別な細胞の「読み替え需要の高さ」と、それにともなうp53を介した細胞死にあることが、近年ようやく解き明かされつつあります。
臨床の現場では、「下あごが小さい」「発達がゆっくり」という個々の所見にとらわれず、進行性の小頭症・特徴的な耳・伝音性難聴・食道閉鎖や心疾患を一つの症候群として束ねて考え、早めにEFTUD2解析やWESへつなぐ鋭さが求められます。とりわけ、見た目が似たトリーチャー・コリンズ症候群や、同じスプライソソーム病でも四肢に異常が出るネイジャー症候群と正確に見分けることは、発達の見通しや内臓奇形のリスク管理に直結する、とても大切な作業です。
根本的な治療はまだ確立されていませんが、新生児期の救命から成人期の社会参加まで、医療・特別支援教育・福祉が一体となった生涯にわたる包括的なケアこそが、ご本人とご家族の生活の質を最大化し、その人の持つ力を引き出す道だと、私たちは考えています。
よくある質問(FAQ)
🏥 希少疾患の診断・遺伝カウンセリングについて
ギオン・アルメイダ型下顎顔面異骨症をはじめとする希少な遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にお問い合わせください。
関連記事
参考文献
- [1] OMIM #610536. Mandibulofacial Dysostosis, Guion-Almeida Type; MFDGA. Johns Hopkins University. [OMIM]
- [2] Lines MA, Hartley T, MacDonald SK, et al. Mandibulofacial Dysostosis with Microcephaly. GeneReviews®. University of Washington. [GeneReviews]
- [3] Lines MA, Huang L, Schwartzentruber J, et al. Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am J Hum Genet. 2012;90(2):369-377. [PubMed]
- [4] Orphanet. Mandibulofacial dysostosis-microcephaly syndrome. ORPHA:79113. [Orphanet]
- [5] Huang L, Vanstone MR, Hartley T, et al. Mandibulofacial Dysostosis with Microcephaly: Mutation and Database Update. Hum Mutat. 2016;37(2):148-154. [PMC5512564]
- [6] MedlinePlus Genetics. EFTUD2 gene. National Library of Medicine. [MedlinePlus]
- [7] MedlinePlus Genetics. Mandibulofacial dysostosis with microcephaly. National Library of Medicine. [MedlinePlus]
- [8] Wang Y, et al. Mandibulofacial dysostosis with microcephaly: An expansion of the phenotype via parental survey. Mol Genet Genomic Med. 2021. [PMC8379904]






