目次

CTNNB1症候群は、第3染色体にあるCTNNB1遺伝子の片方が生まれつきうまく働かなくなる(ハプロ不全)ことで起こる、まれな神経発達症です。正式名称を「痙性両麻痺と視覚障害を伴う神経発達症(NEDSDV)」といい、発達の遅れや知的障害、体の中心はやわらかいのに手足がつっぱるという独特の運動障害、目の網膜の異常などを特徴とします。長く「原因不明の脳性麻痺」とされてきた患者さんの中に、この病気が数多く隠れていたことが、近年わかってきました。
Q. CTNNB1症候群とはどのような病気ですか?まず結論だけ知りたいです
A. CTNNB1という遺伝子が生まれつき片方うまく働かないことで起こる、まれな神経発達症です。発達の遅れや知的障害、体幹はやわらかいのに手足がつっぱる二相性の運動障害、目の網膜の病気(FEVR様)などが特徴です。「原因不明の脳性麻痺」と診断されてきた中に、この病気が多く含まれていたことが近年わかり、独立した病気として再定義されました。
- ➤疾患の定義 → OMIM 615075、Orphanet ORPHA:404473、発症は出生10万人あたり約2.6〜3.2人
- ➤原因 → CTNNB1遺伝子の機能喪失(ハプロ不全)。その多くは新生突然変異
- ➤主な症状 → 発達の遅れ・体幹低緊張と手足の痙縮・小頭症・FEVR様の網膜病変
- ➤診断の鍵 → 重い運動障害なのに脳MRIがほぼ正常という「ギャップ」
- ➤最新の治療 → 世界初の遺伝子治療URBAGEN(2025年12月に初回投与を実施)
1. CTNNB1症候群とは:疾患の定義と歴史
CTNNB1症候群は、第3染色体の短い腕(3p22.1)にあるCTNNB1遺伝子の働きが生まれつき不足することで起こる、常染色体顕性(優性)遺伝形式の希少な神経発達症です。正式な医学名は「痙性両麻痺と視覚障害を伴う神経発達症(Neurodevelopmental disorder with spastic diplegia and visual defects:NEDSDV)」で、国際的な遺伝病データベースOMIMには「OMIM 615075」として、Orphanetには「ORPHA:404473」として登録されています。原因遺伝子の名前をとって「CTNNB1症候群」「CTNNB1関連神経発達症」と呼ばれることが、いまでは一般的になっています。
この病気が「ひとつの独立した病気」として認識されるようになったのは、比較的最近のことです。次世代シーケンサー(NGS)という、遺伝子をまとめて速く調べられる技術が医療現場に広まったことが、最大のきっかけでした。それ以前は、特徴的な運動の問題(体幹はやわらかいのに手足がつっぱる)を持つこのお子さんたちは、長らく「原因不明の脳性麻痺(CP)」という大きなくくりの中にまとめられていました。また、一部の女の子では、重い知的障害と特徴的なくり返し動作から、非定型のレット症候群と誤って診断されることもありました。
ところがNGSの普及によって、こうした患者さんたちの中に、CTNNB1遺伝子の変化を共通して持つ均一なグループがいることが明らかになりました。実際、近年の大規模研究では、CTNNB1の機能喪失型変異は「脳性麻痺の単一遺伝子原因として最も多いもの」と報告されています[5]。これは、「脳性麻痺」という診断の中に、実は遺伝的な原因が隠れていることを示す、重要な発見でした。
💡 用語解説:常染色体顕性(優性)遺伝
「常染色体」とは、性別を決める染色体(X・Y)以外の染色体のこと。「顕性(けんせい/旧称:優性)」とは、ペアになっている2本の遺伝子のうちどちらか1本に変化があるだけで症状が出る遺伝の仕方です。CTNNB1症候群はこの形をとりますが、後で説明するように多くは新生突然変異(親にはない、お子さんで初めて生じた変化)で起こるため、実際に親から子へ受け継がれた例はそれほど多くありません。
発症のしやすさに男女の差はなく、世界全体での発症頻度は出生10万人あたり約2.6〜3.2人と推定されています[8]。中核となる特徴は、(1)広く重い精神運動発達の遅れ、(2)体幹の低緊張と手足の痙縮という二相性の運動障害、(3)小頭症、(4)自閉スペクトラム症(ASD)に似た行動、(5)目の網膜の重い病気(FEVR様)——の5つの柱でとらえると理解しやすくなります。
2. 原因遺伝子CTNNB1と分子メカニズム
CTNNB1遺伝子は、β-カテニン(ベータ・カテニン)というタンパク質の設計図です。このタンパク質は全身のさまざまな細胞でつくられ、生命の土台を支える「2つの顔(デュアルファンクション)」を持っています。この2つの働きが同時に低下することが、CTNNB1症候群が多くの臓器に影響を及ぼす理由です。
💡 用語解説:β-カテニンの「2つの顔」
顔その1(細胞をつなぐ「のり」):皮膚や腸、脳の細胞がバラバラにならないようにつなぎとめる「接着」の部品として働きます。これにより組織がきちんと形を保てます。
顔その2(遺伝子の「スイッチ」):細胞の核に入って特定の遺伝子をオンにし、細胞をふやしたり性質を変えたりする命令を出します。この仕組みは「Wnt(ウィント)シグナル」と呼ばれ、体づくりの根幹を担います。
脳の発達では、このWntシグナルが神経細胞をふやし、適切な配線(シナプス)をつくるのに欠かせません。さらに目では、Wntシグナル(特にNorrin-β-カテニン経路)が網膜の細い血管を端まで伸ばす働きを担っており、ここがうまくいかないと、あとで述べるFEVR様の網膜病変が起こります。
💡 用語解説:ハプロ不全(ハプロふぜん)
人は遺伝子を父・母から1本ずつ、計2本持っています。CTNNB1症候群では、その片方が変化して働かなくなり、β-カテニンの量が正常の約半分に減ります。このように「量が半分になっただけで正常な働きが保てなくなり、症状が出る状態」をハプロ不全といいます。脳の神経細胞が正しい場所に並べず、神経のネットワークがうまく作れなくなることが、発達の問題につながります。
💡 用語解説:機能喪失型/機能獲得型と「同じ遺伝子、正反対の病気」
機能喪失型:遺伝子の働きが弱くなる・なくなる変化です。生まれつき全身の細胞にあると、β-カテニンが足りなくなってCTNNB1症候群になります。
機能獲得型:逆に働きが「暴走」する変化です。これは生まれた後に体の一部の細胞だけで起こることが多く、β-カテニンがたまりすぎて大腸がん・子宮内膜がん・デスモイド腫瘍などのがんの原因になります。同じCTNNB1でも、変化の種類と起こる場所が違えば、まったく別の病気になるのです。
この記事で扱うCTNNB1症候群は、あくまで生まれつきの機能喪失型による神経発達症です。がんで見られる体細胞の機能獲得型とは、変化のタイプも場所もまったく異なる別の話題です。CTNNB1の二面性については、遺伝子そのものを詳しく解説したページもご用意しています。
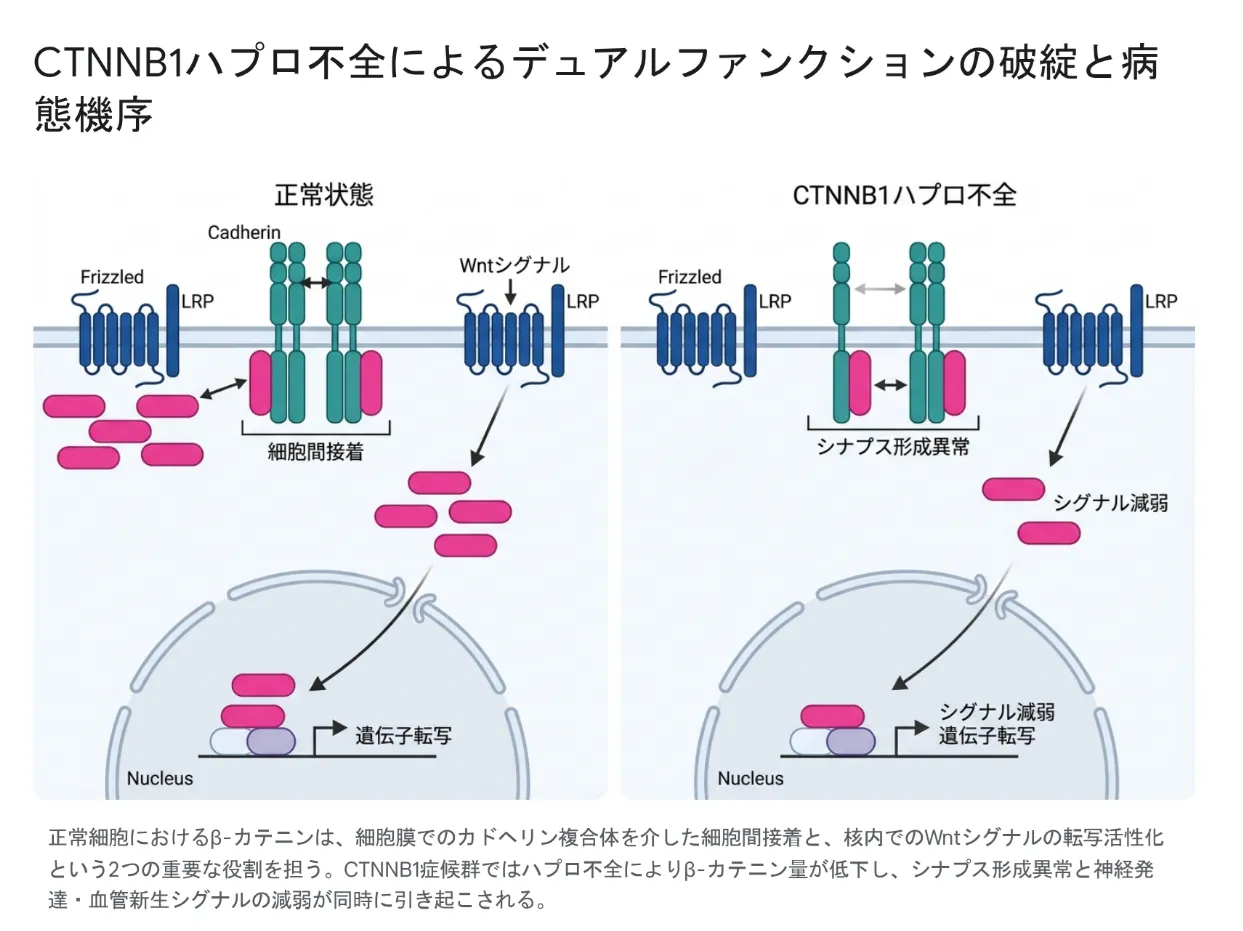
正常な細胞では、β-カテニンは細胞接着(細胞同士のつなぎ)と、核内でのWntシグナルの遺伝子スイッチという2つの役割を担います。CTNNB1症候群ではハプロ不全によってβ-カテニンが不足し、シナプス形成の異常と、神経発達・血管新生シグナルの低下が同時に起こります。
3. 主な症状と特徴
CTNNB1症候群は、変化があれば必ず何らかの症状が出る(浸透率はほぼ完全)一方で、症状の種類や重さの個人差がとても大きいことが知られています。それでも、多くの患者さんに共通してみられる中核症状があります。まずは主な症状の出現頻度を見てみましょう。
CTNNB1症候群でみられる主な症状の出現頻度
発達の遅れや筋緊張の異常はほぼ全例にみられる中核症状です。一方、赤色のFEVR様網膜病変や脊髄係留症候群は頻度こそ中程度ですが、放置すると失明や運動機能の低下など取り返しのつかない後遺症につながるため、症状がなくても定期的なチェックがすすめられます。データ出典:GeneReviews(NBK580527)[3]ほか。
発達・知能・ことばの特徴
ほぼすべての患者さんに、軽度から重度までの全体的な発達の遅れと知的障害がみられます。ことばの遅れはとくに目立ち、約97%に認められます。一方で、自分から話す力(表出)に比べて、相手の言葉を理解する力(受容)は比較的保たれている傾向があります。そのため、声でうまく話せなくても、手話やジェスチャー、絵カード、タブレットなどの拡大代替コミュニケーション(AAC)を使えば、豊かに気持ちを伝え合えるお子さんが多くいます。また、成長とともに認知・適応の力がゆっくりでも着実に伸びていく傾向も報告されており、継続的な療育がとても大切です[7]。
「やわらかい体幹」と「つっぱる手足」——二相性の運動障害
この病気を最も特徴づける身体のサインが、運動の問題です。赤ちゃんの時期は首や体幹の力が弱く(体幹低緊張、フロッピーインファント様)、首すわりやおすわりが遅れます。ところが成長とともに、手足、とくに下肢の筋肉が強くつっぱる(痙縮)ようになります。この「中心はやわらかいのに末端はつっぱる」二相性のパターンが、下肢中心の痙性両麻痺をつくり、脳性麻痺と見分けがつきにくい原因になります。つま先立ち歩き(尖足歩行)や、両脚が交差するはさみ足歩行がみられることもあります。このほか、ジストニア(筋肉の異常な収縮)や運動失調、過剰な驚愕反応なども報告されています。
💡 用語解説:痙縮(けいしゅく)と痙性両麻痺
痙縮とは、筋肉が異常に緊張してつっぱり、動かしにくくなる状態です。手足の関節を曲げ伸ばししようとすると強い抵抗があります。痙性両麻痺とは、この痙縮が主に両方の脚(下肢)に出るタイプを指します。CTNNB1症候群では加齢とともにこの痙縮が進むことが多く、放っておくと関節が固まる(拘縮)ため、早めのリハビリと装具が役立ちます。
目の合併症:失明を防ぐために最重要
目の異常は、学習や生活の質に大きく影響する、見逃せない要素です。約39%の患者さんに、家族性滲出性硝子体網膜症(FEVR)に似た重い網膜血管の異常が認められます。Wntシグナルが弱まることで、網膜の周辺部まで血管が伸びきらず(無血管化)、虚血になった部分から異常な新生血管が増えて、線維化・牽引・網膜剥離へと進むことがあります。進行すると不可逆的な失明につながる恐れがあるため、症状がなくても定期的な眼科チェックが欠かせません。このほか、斜視(約55%)や近視・遠視・乱視などの屈折異常(約27%)もよくみられます。
行動・睡眠・てんかん、そして見逃しやすい合併症
視線が合いにくい、手をもむような常同運動、こだわりなどASD様の特性が約34〜46%に、注意欠如・多動(ADHD)様の特性も一部にみられます。性格は明るく人懐っこいと表現されることが多い一方で、不安が強かったり、かんしゃくや自傷がみられることもあり、ご家族の負担になることがあります。睡眠の問題も約35%に。てんかんの合併は他の重い神経発達症に比べると少なめ(約10〜12%)で、難治化することはまれです。
とくに注意したいのが、約25%にみられる脊髄係留症候群です。これは脊髄の下端が脊柱管の中で異常に癒着し、成長に伴って引っぱられて傷つく状態です。下肢の痙縮の急な悪化や排尿・排便の異常として現れますが、CTNNB1症候群はもともと下肢の痙縮が進むため、「もとの病気の自然な経過」と誤解されて見逃されやすい点が落とし穴です。下肢症状が急に進む場合は、脊髄MRIでの精査が強くすすめられます。

4. 鑑別診断:脳性麻痺・レット症候群との違い
CTNNB1症候群は、いくつかの古典的な病気と症状が大きく重なるため、慎重な見分けが必要です。とくに重要なのは、「重い運動障害があるのに、脳MRIがほぼ正常」という特徴的なギャップです。
💡 CTNNB1症候群を疑うべき手がかり
- ➤仮死・超早産・重症黄疸など、周産期の明らかな異常の病歴がない
- ➤脳MRIに運動障害を説明できるほどの大きな脳損傷が見当たらない
- ➤それでも体幹低緊張+下肢の痙縮が進行する
- ➤ことばの遅れやFEVR様の網膜病変を伴う
一般的な脳性麻痺では、頭部MRIに脳室周囲白質軟化症(PVL)などの器質的な傷あとが見つかるのが普通です。ところがCTNNB1症候群では、MRIが「完全に正常」か、脳梁の低形成・軽い髄鞘化の遅れ・側脳室の軽い拡大など、症状の重さに比べてごくわずかな所見にとどまることがほとんどです。この「画像所見のパラドックス」に気づけるかどうかが、正しい遺伝子検査への近道になります。主な鑑別疾患を整理します。
脳性麻痺(CP)との鑑別
脳性麻痺は症状をまとめた呼び名であり、周産期の脳障害の病歴やMRIの器質的病変が手がかりです。原因不明のCPに遺伝子解析を行うと、一定割合でCTNNB1変異が見つかる「遺伝性CPミミック」である点に最大の注意が必要です。
レット症候群との鑑別
とくに女の子では、手をもむ常同運動・重い知的障害・呼吸の異常など、MECP2遺伝子によるレット症候群とそっくりな像(Rett様)を示すことがあります。症状だけでは見分けがつきにくく、遺伝子検査での区別が必須です。
網膜の病気を主とする疾患
孤発性のFEVR(FZD4・LRP5・NDP・TSPAN12など)やKIF11異常も網膜病変を示しますが、CTNNB1症候群のような重い発達遅滞・小頭症・体幹低緊張+痙縮を伴うことはまれで、全身の中枢神経症状の有無が決め手になります。
5. 診断と遺伝子検査の進め方
CTNNB1症候群には特有の血液マーカーがないため、確定診断は遺伝学的検査に完全に頼ります。ここで大切なのは、「生まれた後の検査」と「生まれる前の検査」をはっきり分けて考えることです。検査=出生前、という誤解を避けましょう。
出生後(生まれた後)の確定診断
お子さんの症状からCTNNB1症候群が疑われる場合は、血液や唾液などを用いた遺伝子検査でCTNNB1を調べます。現在の標準は、全エクソーム解析(WES)・全ゲノム解析(WGS)、または神経発達症をまとめて調べる遺伝子パネル検査です。これらで、CTNNB1の中に生じた一塩基の置き換えや、数塩基の欠失・挿入などの小さな変化を正確に見つけます。
一方、数パーセントの患者さんでは、CTNNB1遺伝子そのものではなく、その遺伝子を含む3p22.1領域の微細な欠失が原因です。この場合は、通常の染色体検査(Gバンド法)では見つけにくいため、マイクロアレイ染色体検査(CMA)が威力を発揮します。
💡 用語解説:ミスセンス変異・ナンセンス変異・フレームシフト変異
ミスセンス変異:DNAの文字が1つ変わり、タンパク質を作る部品(アミノ酸)が別のものに置き換わる変化です。
ナンセンス変異:本来より早い位置に「ここで終わり」の合図ができてしまい、短い不完全なタンパク質になる変化です。
フレームシフト変異:文字が抜けたり入ったりして「3文字ずつ読むルール」がずれてしまう変化です。多くはうまく働かないタンパク質になります。CTNNB1症候群では、こうした機能喪失型の変化が原因になります。
出生前(生まれる前)の検査
妊娠中にCTNNB1を含む多数の遺伝子を調べる選択肢として、当院のNIPT(新型出生前診断)のインペリアルプランがあり、CTNNB1はその対象遺伝子に含まれています。ただしNIPTはあくまでスクリーニング(ふるい分け)であり、確定診断ではありません。出生前により確実な診断が必要な場合の確定検査は、羊水検査・絨毛検査です。羊水・絨毛で採取した細胞にCMAを行えば、Gバンド法では見つからない微細欠失も確定的に調べられます。
日本での診断基準と難病・福祉制度
日本でもゲノム医療が普及し、原因不明とされていた発達遅滞・脳性麻痺の中からCTNNB1症候群と確定診断される例が増えています。患者さんが医療費助成や福祉・教育の支援を受けやすくするため、研究班によって「CTNNB1関連神経発達症(3p22.1欠失症候群)」として診断基準の整備が進められています[11]。現在、国の指定難病・小児慢性特定疾病への認定に向けた要件づくりが行われている段階です。
| 区分 | 内容 |
|---|---|
| 大症状 | I. 知的発達症(IQ70未満)または年齢に不相応な発達遅滞(必須項目)/ II. 四肢の筋痙縮(末梢の痙性麻痺)/ III. 滲出性硝子体網膜症(FEVR様所見) |
| 小症状 | I. 小頭症/ II. 大症状以外の眼科所見(斜視・近視や遠視などの屈折異常)/ III. 行動異常(ASD・ADHD・常同運動など) |
| 確定診断 | 大症状I(必須)を認め、かつ遺伝学的検査でCTNNB1の機能喪失型病的バリアント、または3p22.1領域の欠失が証明された場合 |
6. 治療と長期管理
現時点では、失われたβ-カテニンを直接補って病気を根本から治す承認薬はありません。そのため治療の中心は、生活の質(QOL)を最大限に高め、二次的な合併症を予防する集学的な対症療法です。小児神経科・整形外科・眼科・リハビリ科(理学療法士・作業療法士・言語聴覚士)などが連携するチーム医療が欠かせません。
リハビリテーション(療育)
理学療法(PT)
体幹の低緊張に対しては、姿勢を保つための筋力とバランスの訓練を。手足の痙縮に対しては、関節が固まる(拘縮)のを防ぐストレッチと関節可動域訓練を毎日くり返し、移動手段の獲得を目指します。
作業療法(OT)
腕は上がっても、細かい手先の操作(巧緻運動)が苦手なお子さんが多いため、遊びを通じた手先の訓練を行います。食事・着替え・排泄など、日常生活動作の自立も支援します。
言語聴覚療法(ST)
理解力が保たれている強みを活かし、無理に発声だけを求めず、絵カードやタブレットなどのAAC機器を積極的に導入。お子さんがフラストレーションなく気持ちを伝えられる手段づくりをサポートします。
痙縮・運動障害への対応
進行する下肢の痙縮には、年齢と重症度に応じて段階的に対応します。足首のつっぱりを抑える短下肢装具(AFO)、全身の緊張をやわらげる内服薬(バクロフェンなど。ただし体幹低緊張を悪化させうるため慎重に調整)、特定の筋肉に注射するボツリヌス療法、そして拘縮が強い場合の腱延長などの整形外科手術があります。前述の脊髄係留症候群が原因と判断された場合は、脳神経外科による解除術(終糸離断術)が検討されます。適切なタイミングでの介入が、運動機能を守るうえで重要です。
目の合併症への先制的なケア
最大の脅威であるFEVR様病変は、通常の眼底検査だけでは周辺部の異常を見落とすことがあります。そのため、必要に応じて蛍光眼底造影検査による定期スクリーニングが推奨されます。早期に無血管領域や新生血管が見つかれば、レーザー光凝固術で網膜剥離への進行を食い止められる可能性があります。斜視や屈折異常には、乳幼児期から眼鏡やアイパッチで弱視を予防します。
最新の治療研究:世界初の遺伝子治療「URBAGEN」
対症療法しかなかったこの病気に、いま根本原因に直接はたらきかける遺伝子補充療法が登場しつつあります。スロベニアを拠点とする非営利のCTNNB1財団が主導して開発した、世界初のCTNNB1症候群向け遺伝子治療薬候補が「URBAGEN(ウルバジェン)」です[9]。
💡 用語解説:AAV9ベクターと脳室内投与(ICV)
AAV9ベクター:病原性をなくした安全なウイルスの殻を「運び屋」として使い、正常なCTNNB1遺伝子を神経細胞に届ける仕組みです。脊髄性筋萎縮症(SMA)の治療薬でも実用化されており、ゲノムに組み込まれにくく発がんリスクが低いという利点があります。
脳室内投与(ICV):薬を血管ではなく、脳の中心にある脳脊髄液の空間(脳室)に直接注入する方法です。血液脳関門を迂回し、少ない量で脳全体に効率よく届けられます。
URBAGENは、動物モデルで歩行などの改善が確認され、2025年7月には欧州医薬品庁(EMA)から希少疾病用医薬品(オーファンドラッグ)に指定されました。そして、スロベニアのリュブリャナ大学医療センターを実施施設とする第I/II相臨床試験「GAIN-CTNNB1試験」(NCT07270549)が始まり、2025年12月10日には、世界で初めて1人の患者さんへの投与が行われました[10]。今後、2〜12歳の合計12名(多くは国外から)に順次投与し、5年かけて安全性と効果を確認していく計画です。
ただし、これは現在まさに臨床試験が進んでいる段階であり、誰でもすぐに受けられる確立した治療ではありません。期待が大きい分、最新情報を正しく理解し、いま目の前のお子さんに必要なケアを着実に積み重ねることが何より大切です。
7. 遺伝カウンセリングの意義
CTNNB1症候群に関わる遺伝子検査では、結果の受け止め方やこれからの見通しについて、専門家とゆっくり話し合う遺伝カウンセリングが大切です。主に次のような内容を扱います。
- ➤遺伝の仕方と再発の可能性:多くは新生突然変異で、ご両親には変異が見られないことがほとんどです。ただし常染色体顕性(優性)遺伝のため、ご本人がお子さんを持つ場合に伝わる確率は理論上50%です。親の一部の細胞だけに変異がある「生殖細胞モザイク」の可能性もゼロではないため、次のお子さんを考える際の相談が役立ちます。
- ➤結果の意味づけ:同じCTNNB1でも、生まれつきの機能喪失型(症候群)と、がんで見られる体細胞の機能獲得型はまったくの別物です。混同を避ける丁寧な説明が必要です。
- ➤検査の選択肢:必要に応じて羊水検査・絨毛検査などの確定検査について、利点と限界の両面から中立的にご説明します。
- ➤心理的サポート:まれな病気ゆえに情報が限られるため、不安や疑問に寄り添いながら、長期的に医療とつながり続けられるよう支えます。
遺伝カウンセリングは、臨床遺伝専門医をはじめとする専門家が担います。決定を急かすのではなく、ご家族が納得して選べるよう伴走することを大切にしています。
8. よくある誤解
誤解①「脳性麻痺だから遺伝とは無関係」
脳性麻痺と診断されていても、原因不明のケースにはCTNNB1のような遺伝的原因が隠れていることがあります。MRIが正常に近いのに重い運動障害がある場合は、遺伝子検査を考える価値があります。
誤解②「両親が健康だから遺伝子の病気ではない」
CTNNB1症候群の多くは新生突然変異で、ご両親には変異がないことがほとんどです。「両親が健康=遺伝子の病気ではない」という思い込みが、診断を遅らせることがあります。
誤解③「CTNNB1の変異=がんになる」
がんで見つかるCTNNB1変異の多くは、生まれた後に腫瘍の細胞だけで起こる体細胞変異です。生まれつきの機能喪失型(症候群)があるからといって、がんになりやすいわけではありません。
誤解④「もう治療法が完成している」
遺伝子補充療法(URBAGEN)の研究は大きく前進し初回投与も行われましたが、いまは臨床試験の段階です。誰でも受けられる確立した治療ではない点に注意が必要です。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 希少疾患の診断・遺伝カウンセリングについて
CTNNB1症候群をはじめとする希少遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] OMIM #615075. Neurodevelopmental Disorder with Spastic Diplegia and Visual Defects (NEDSDV). Johns Hopkins University. [OMIM]
- [2] OMIM *116806. Catenin Beta-1; CTNNB1. Johns Hopkins University. [OMIM]
- [3] GeneReviews. CTNNB1 Neurodevelopmental Disorder. NCBI Bookshelf (NBK580527). [GeneReviews]
- [4] Orphanet. Neurodevelopmental disorder with spastic diplegia and visual defects. ORPHA:404473. [Orphanet]
- [5] Kayumi S, et al. Genomic and phenotypic characterization of 404 individuals with neurodevelopmental disorders caused by CTNNB1 variants. Genet Med. 2022;24(11):2351-2366. [Genetics in Medicine]
- [6] Lee S, et al. The extended clinical and genetic spectrum of CTNNB1-related neurodevelopmental disorder. Front Pediatr. 2022;10:960450. [PubMed]
- [7] Sudnawa KK, et al. Clinical Phenotypic Spectrum of CTNNB1 Neurodevelopmental Disorder. PMC11872165. 2024. [PMC]
- [8] Zhuang W, et al. CTNNB1 in neurodevelopmental disorders. Front Psychiatry. 2023;14:1143328. [PMC]
- [9] CTNNB1 Foundation. Gene Replacement Therapy Program (URBAGEN). [CTNNB1 Foundation]
- [10] ClinicalTrials.gov. Gene Replacement Therapy for Paediatric Patients With CTNNB1 Neurodevelopmental Syndrome (GAIN-CTNNB1 / NCT07270549). [ClinicalTrials.gov]
- [11] CTNNB1関連神経発達症(3p22.1欠失症候群)診断基準. マイクロアレイ染色体検査研究班. [研究班ページ]






