目次

反復発作性失調症2型(EA2)は、数時間から数日続く激しいめまいと体幹失調の発作を繰り返す遺伝性のチャネル病です。第19番染色体上のCACNA1A遺伝子変異により、神経細胞のカルシウム流入を担うP/Q型カルシウムチャネルの機能が低下することで発症します。アセタゾラミドや4-アミノピリジン(ファンプリジン)による予防治療で発作の頻度を大きく減らすことが可能であり、早期診断と適切な薬物治療が患者さんの生活の質を大きく改善します。
Q. 反復発作性失調症2型(EA2)はどんな病気ですか?まず結論を教えてください
A. CACNA1A遺伝子の変異によって起こる、常染色体顕性遺伝のチャネル病(イオンチャネル機能異常による疾患)です。数時間から数日にわたる激しいめまい、ふらつき、ろれつが回らないといった発作を繰り返し、運動やストレスで誘発されます。アセタゾラミドや4-アミノピリジンによる予防治療が有効で、適切に管理すれば発作の頻度と重症度を大幅に減らせます。
- ➤疾患の定義 → OMIM #108500、Orphanet ORPHA:97、推定有病率10万人に1人未満
- ➤原因遺伝子 → CACNA1A(19p13)、P/Q型カルシウムチャネルCav2.1の機能喪失
- ➤主な症状 → 数時間〜数日続くめまい・体幹失調・構音障害、発作間欠期の眼振
- ➤鑑別診断 → EA1・前庭性片頭痛・脊髄小脳変性症6型(SCA6)との見分け方
- ➤治療 → アセタゾラミド(奏効率約70%)、4-AP/ファンプリジン(RCTで発作63%減)
1. 反復発作性失調症2型(EA2)とは:疾患の基本概念
反復発作性失調症(Episodic Ataxia: EA)は、体のバランスが取れなくなる「失調」の発作を繰り返す遺伝性疾患グループの総称です。現在までに少なくとも11種類のサブタイプが知られていますが、その中で最も頻度が高く、臨床現場で最もよく遭遇するのが反復発作性失調症2型(EA2)です。推定有病率は10万人に1人未満と非常に稀な疾患ですが、遺伝子検査の難しさから実際の患者数はもっと多い可能性があると考えられています。
💡 用語解説:常染色体顕性遺伝(じょうせんしょくたいけんせいいでん)
「常染色体」とは性染色体(X・Y)以外の染色体のこと。「顕性(けんせい)」は以前「優性(ゆうせい)」と呼ばれていた遺伝形式で、ペアになった染色体のどちらか1本に変異があるだけで症状が現れる遺伝の仕方を指します。EA2では、両親のどちらかからこの変異を受け継ぐと発症します。患者さん本人が子どもを持つ場合、その子どもへの遺伝確率は理論上50%です。
EA2の臨床像を一言で表すなら、「数時間〜数日にわたる激しいめまいと体幹失調の反復発作」です。発作の引き金となるのは、激しい運動や精神的ストレス、カフェイン、アルコール、発熱など日常的に遭遇する要素であり、患者さんの社会生活に大きな影響を及ぼします。さらに、発作と発作の間(発作間欠期)にも特徴的な眼球運動異常が持続し、長期的には小脳の萎縮による慢性的な失調が進行する場合もあります。
💡 用語解説:チャネル病(チャネロパチー)
細胞膜に存在する「イオンチャネル」(ナトリウム・カリウム・カルシウムなどのイオンを細胞内外に出し入れする穴のような構造)の機能異常によって起こる病気の総称です。神経細胞や筋肉細胞はイオンの流れによって電気信号を生み出すため、チャネルが正しく働かないと、てんかん・麻痺・不整脈・運動失調など多彩な症状が現れます。EA2はカルシウムチャネルのチャネル病に分類されます。
2. 原因遺伝子CACNA1Aと分子メカニズム
EA2の原因は、第19番染色体短腕(19p13)にあるCACNA1A遺伝子の変異です。この遺伝子について、より詳しくはCACNA1A遺伝子の解説ページもあわせてご覧ください。
💡 用語解説:P/Q型電位依存性カルシウムチャネル(Cav2.1)
CACNA1Aがコードするタンパク質は「Cav2.1(キャブ・ツー・テン)」または「α1A(アルファ・ワン・エー)サブユニット」と呼ばれる、P/Q型カルシウムチャネルの主要な部品です。このチャネルは中枢神経系全般に存在しますが、とくに小脳のプルキンエ細胞と顆粒細胞で非常に多く発現しています。神経細胞に電気信号が届くと、このチャネルが開いてカルシウムイオンが流入し、シナプスでの神経伝達物質の放出を引き起こします。さらに、小脳プルキンエ細胞のリズミカルな自発発火(ペースメーカー機能)を支える重要な役割も担っています。
EA2を引き起こす「機能喪失型変異」
EA2の患者さんでは、これまでに30種類以上の異なるCACNA1A遺伝子変異が報告されています。その大部分は「機能喪失型(Loss-of-Function: LOF)」と呼ばれるタイプで、ナンセンス変異・スプライス部位変異・微小欠失や挿入によるフレームシフトなどが含まれます。これらの変異は途中で切れた不完全なタンパク質を生み出すか、あるいはタンパク質の発現量そのものを低下させることで、細胞膜上の機能的なカルシウムチャネルの数を減少させます(ハプロ不全)。
💡 用語解説:ハプロ不全(ハプロインサフィシエンシー)
ヒトの遺伝子は通常、父親由来と母親由来の2本ペアで存在し、両方が機能することで「定員」を満たしています。片方の遺伝子が機能しなくなり、もう片方だけでは細胞が必要とする量に達しない状態を「ハプロ不全」と呼びます。EA2では、機能喪失型変異を持つ遺伝子から十分なカルシウムチャネルが作られず、結果として神経細胞のカルシウム流入が不足します。
「1つの遺伝子、複数の疾患」:CACNA1A関連疾患のスペクトラム
CACNA1A遺伝子は、変異の種類によって全く異なる3つの神経疾患を引き起こすことで知られています。同じ遺伝子なのに表現型が大きく異なる「一つの遺伝子、複数の疾患(One gene, multiple diseases)」の典型例です。
| 疾患名 | 変異の種類 | 主な特徴 |
|---|---|---|
| 反復発作性失調症2型 (EA2) |
点突然変異・微小欠失・挿入 (機能喪失型) |
小児期〜若年発症。数時間〜数日続く反復発作性めまい・失調。発作間欠期の眼振 |
| 家族性片麻痺性片頭痛1型(FHM1) | ミスセンス変異 (機能獲得型) |
一側性の運動麻痺(片麻痺)を伴う重篤な前兆付き片頭痛 |
| 脊髄小脳変性症6型(SCA6) | CAGリピート異常伸長 (ポリグルタミン病) |
成人期後期(50代以降)発症。緩徐進行性の小脳失調・構音障害・眼振 |
家族性片麻痺性片頭痛1型(FHM1)は「機能獲得型(Gain-of-Function)」変異によって引き起こされ、チャネルの開口確率が上昇して脳内が過興奮状態になることで重篤な片頭痛を生じます。一方、脊髄小脳変性症6型(SCA6)は遺伝子末端のCAG(シーエージー)リピート配列が異常に伸長することで、毒性を持つ異常タンパク質がプルキンエ細胞に蓄積する「ポリグルタミン病」の一種です。
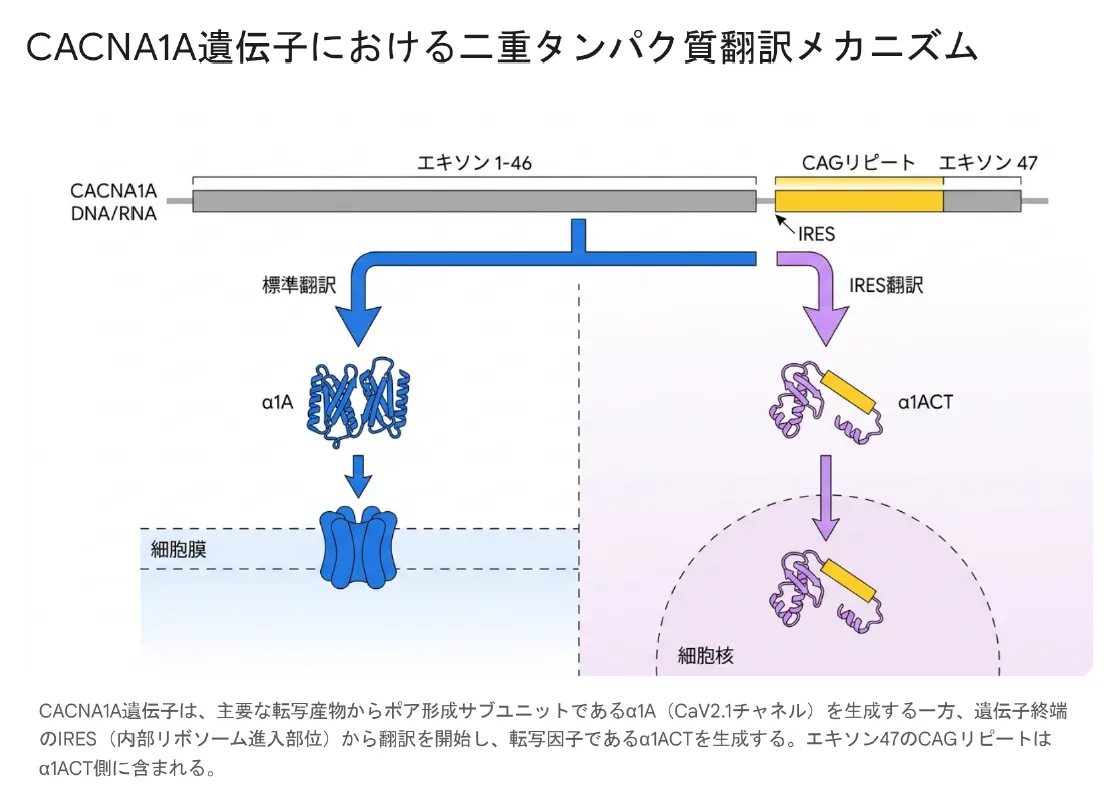
CACNA1A遺伝子は同一のmRNAから2種類の機能タンパク質(カルシウムチャネル本体と転写因子α1ACT)を産生する複雑な構造を持ち、変異の部位と種類によって異なる臨床表現型を生じます。
興味深いことに、これらの疾患の表現型は厳密に分かれているわけではなく、しばしば重なります。EA2患者さんの約50%は何らかの片頭痛を合併し、その一部は片麻痺性片頭痛の基準を満たします。SCA6の初期症状が反復発作性失調として現れることもあり、FHM1家系の約40〜50%で眼振や軽度の進行性失調といった小脳症状が観察されます。
3. 主な症状と進行のプロファイル
EA2の症状は、激しい発作が起こる「発作期」と、それ以外の「発作間欠期」、そして長期経過に伴う進行性変化に分けて理解する必要があります。
発作期:突然襲う激しいめまいと失調
EA2の発作は急激に始まり、数分から数時間、場合によっては数日間にわたって続きます。この「発作の持続時間が長い」という特徴は、後述するEA1との重要な区別ポイントとなります。
🌀 中核症状
- 空間識を失うほどの重度の中枢性めまい
- 体幹の不安定性・歩行不能レベルの体幹失調
- 重症時は車椅子使用や床への座り込みが必要
- 激しい回転性めまいと嘔吐
😵 随伴症状
- 構音障害(ろれつが回らない)
- 複視(物が二重に見える)
- 耳鳴り・全般的な筋力低下
- 悪心・嘔吐
- 一部で片麻痺・ジストニア様姿勢
⚡ 発作の誘発要因
- 激しい身体的労作(運動)
- 精神的ストレス
- カフェイン・アルコール摂取
- 発熱・環境的な暑さ
- 睡眠不足・特定の薬剤
⏱ 発作の特徴
- 持続時間:数時間〜数日
- 急激な発症
- 発症年齢の中央値:約10歳
- 頻度:個人差が大きい
発作間欠期:発作がない時にも残る特徴的サイン
EA2患者さんの大きな特徴のひとつは、発作がない時期にも持続する眼球運動異常です。これは診断の強力な手がかりとなります。
💡 用語解説:下向き眼振・注視方向性眼振
眼振(がんしん)とは、自分の意思とは関係なく眼球が小刻みに揺れる現象です。下向き眼振は眼球が下方向へ振れるタイプで、注視方向性眼振は左右の端を見たときに見ている方向に向けて眼球が振れるタイプを指します。どちらも小脳機能の異常を示唆する「中枢性」の眼振であり、内耳から来る末梢性のめまいとは区別される重要な所見です。EA2では発作のない時期にもこれらの眼振が観察され、診断の決め手のひとつとなります。
また、EA2では筋波動症(ミオキミー)が見られないことも特徴です。これはEA1(次のセクションで詳述)との重要な鑑別点になります。EA2における筋肉の症状としては、ピクつきよりもむしろ筋肉の硬直やジストニア(持続的な異常姿勢)が観察されることがあります。
長期的な進行:小脳萎縮と慢性失調
EA2は「発作性」の疾患として定義されていますが、長期経過に伴い発作間欠期にも持続する進行性の小脳失調を呈する患者さんが多いことが分かっています。MRI検査では、小脳虫部(小脳の正中部分)を中心とした軽度から中等度の小脳萎縮が一般的に観察されます。度重なる発作による神経細胞へのストレスや、長期間のカルシウムチャネル機能不全が、最終的にプルキンエ細胞の不可逆的な細胞死を引き起こすと考えられています。
神経発達・精神医学的合併症
次世代シーケンサー(NGS)技術の発展により、CACNA1A変異は古典的なEA2の枠を超えて、広範な小児期の神経発達障害に関与することが明らかになってきました。とくに小児期発症のEA2では、以下の合併症が報告されています。
・全体的発達遅滞(GDD)・知的障害
・自閉スペクトラム症(ASD)
・体幹筋緊張低下(Hypotonia)
・てんかん(最大62%):小児欠神てんかんから難治性てんかん症候群まで多様
・行動の調節不全
重症例では、生後早期に発症する発達性てんかん性脳症42型(DEE42)と診断されることもあり、CACNA1A変異の臨床スペクトラムは極めて広範であることが分かります。
4. 鑑別診断:EA1・前庭性片頭痛との見分け方
EA2は、症状が似た他の疾患と誤診されやすい疾患です。とくに重要な鑑別対象はEA1と前庭性片頭痛の2つです。
1型反復発作性失調症(EA1)との鑑別
EA2の最重要鑑別疾患は、第12番染色体のKCNA1遺伝子変異(カリウムチャネルKv1.1)に起因するEA1(1型反復発作性失調症)です。両者はどちらも反復性の失調を呈しますが、治療に対する反応が異なるため、早期の鑑別が極めて重要となります。
📊 EA1とEA2の臨床的特徴比較
運動誘発性トリガーの有病率
毎日の発作頻度の有病率
EA1は急激な動作で誘発されやすく、発作頻度が高い傾向にあります。EA2は誘発因子が多様で、発作頻度は比較的低い一方、1回の発作持続時間が長いという特徴があります。
💡 用語解説:筋波動症(ミオキミー / Myokymia)
顔面や四肢の筋肉が持続的に細かくピクピクと波打つように動く現象です。「ミミズが皮膚の下を這うような」と表現されることもあります。EA1患者さんではこの筋波動症が発作のない時期にも観察されることが多く、その特異度は約99.6%と極めて高い診断指標となります。一方、EA2では通常この症状は見られません。
最近のシステマティックレビューと統計解析に基づく診断アルゴリズムによれば、以下の特徴で両者を高い精度で区別できます。
| 特徴 | EA1(KCNA1変異) | EA2(CACNA1A変異) |
|---|---|---|
| 発作の持続時間 | 数秒〜数分(10分未満:感度75.3%・特異度94.0%) | 数時間〜数日 |
| 発症年齢の中央値 | 7歳(より早期) | 10歳 |
| 発作間欠期の筋波動症 | あり(特異度99.6%) | なし |
| 発作間欠期の眼振 | 通常なし | あり(特異度98.8%) |
| 第一選択薬 | 特定の抗てんかん薬(カルバマゼピン等) | アセタゾラミド・4-アミノピリジン |
前庭性片頭痛(Vestibular Migraine)との鑑別
EA2は前庭性片頭痛(VM)と誤診されやすい疾患の代表格です。EA2患者さんの約50%は片頭痛を合併し、発作性の回転性めまいを呈するため、初期の臨床症状だけではVMと見分けることが非常に困難なケースがあります。実際に、運動誘発性の重度めまいと頭痛を反復していた患者さんが当初「片頭痛の亜型」と誤診され、後に詳細な再評価でEA2と判明した事例も報告されています。
鑑別の決め手:VM患者さんは通常、発作のない時期にこのような持続的な中枢性の小脳・眼球運動障害を示しません。発作間欠期の注視方向性眼振や進行性の小脳症状の有無が、最も信頼性の高い鑑別指標となります。
そのほかの鑑別対象として、メニエール病・良性発作性頭位めまい症(BPPV)・前庭神経炎などの末梢性前庭疾患、多発性硬化症などの脱髄性疾患、脳卒中、傍腫瘍性小脳変性症、グルテン失調症などが挙げられます。これらは神経学的診察と画像検査によって除外していきます。

5. 診断と遺伝子検査の進め方
EA2の診断は、詳細な病歴聴取・神経学的診察・遺伝子検査の3つの組み合わせによって確立されます。とくに発作の持続時間・誘発要因・発作間欠期の所見を体系的に評価することが、適切な治療方針の決定に不可欠です。
臨床的レッドフラッグ:EA2を強く疑うべきサイン
💡 EA2を強く疑う臨床所見の組み合わせ
- ➤数時間〜数日続く反復性のめまい・体幹失調発作
- ➤発作間欠期の下向き眼振または注視方向性眼振
- ➤運動・ストレス・カフェイン・発熱などの誘発要因
- ➤類似症状の家族歴(常染色体顕性遺伝のため)
- ➤MRI上の小脳虫部の萎縮(長期経過例)
分子遺伝学的検査:確定診断はCACNA1A遺伝子解析
臨床的にEA2が強く疑われる場合、確定診断は遺伝子検査によって行われます。具体的には、CACNA1A遺伝子の全エクソンおよびその近傍のイントロン配列に対する直接シーケンス解析、ターゲット遺伝子パネル、あるいは全エキソームシーケンス(WES)が用いられます。
💡 用語解説:de novo変異(デノボ変異・新生変異)
両親の生殖細胞(精子・卵子)または受精直後に新しく生じた変異で、両親には同じ変異が存在しません。「常染色体顕性遺伝=両親のどちらかが発症している」とは限らず、EA2では家族歴のない孤発例であってもde novo変異によって発症することがあります。家族歴がないからといってEA2を診断から除外することはできません。
遺伝子検査でできること
遺伝子検査によって変異の種類(CAGリピート伸長か、ミスセンス変異か、機能喪失型の点突然変異か)を特定することは、以下の点で大きな意味を持ちます。
- ➤診断の確定:類似疾患(EA1、前庭性片頭痛、SCA6、FHM1)と明確に区別できます
- ➤予後予測:変異の種類によって発症年齢・症状の重症度・合併症リスクが異なります
- ➤治療方針の決定:てんかん合併例では変異プロファイルに応じた抗てんかん薬選択が可能
- ➤家族への情報提供:正確な再発リスクに基づく遺伝カウンセリングが可能になります
🔬 ミネルバクリニックでの検査体制
当院では、CACNA1Aを含む154遺伝子・218疾患を網羅するインペリアルプランでの出生前スクリーニングが可能です。すでに家族内にCACNA1A変異が同定されている場合の出生前確定診断としては、絨毛検査・羊水検査での標的変異解析が選択肢となります。詳細は臨床遺伝専門医へご相談ください。
6. 治療と長期管理:薬物療法と生活指導
EA2の管理は「予防的薬物療法」「トリガー回避を中心とした生活指導」「進行性症状へのリハビリテーション」の3本柱から成り立ちます。とくに薬物療法では、長年の第一選択薬と新しい治療薬がそれぞれ重要な役割を担っています。
第一選択薬:アセタゾラミド(Acetazolamide)
炭酸脱水酵素阻害薬であるアセタゾラミドは、長年にわたりEA2の第一選択薬として用いられてきた薬剤です。臨床的にはEA2患者さんの約70%が奏効し、発作の頻度と重症度が劇的に減少します。
💡 用語解説:アセタゾラミドの作用機序
脳内の炭酸脱水酵素を阻害することで細胞内および脳脊髄液のpHを酸性に傾けます。このpH変化が神経細胞の経膜電位を変化させ、過興奮状態のイオンチャネルを安定化し、発作の閾値を上昇させると考えられています。正確な作用機序は完全には解明されていませんが、偶然の発見から長年の臨床経験で有効性が確立された薬剤です。
注意すべき副作用:長期服用に伴う副作用が問題となります。四肢の感覚異常(チクチクするしびれ感)、味覚障害、胃腸障害、疲労感、腎結石のリスク増加などが報告されており、ランダム化比較試験ではアセタゾラミド投与群の44.9%で何らかの有害事象が観察されています。また、効果が時間とともに減弱する症例も少なくありません。
画期的新薬:4-アミノピリジン(4-AP)/ファンプリジン
近年、EA2の病態生理学の核心である「プルキンエ細胞のペースメーカー機能低下」に直接アプローチする画期的な治療薬として確立されたのが、カリウムチャネル阻害薬の4-アミノピリジン(4-AP)と、その徐放性製剤であるファンプリジン(Fampridine / Dalfampridine)です。
💡 用語解説:4-AP/ファンプリジンの作用メカニズム
4-APは、Kv1.5サブタイプなどの電位依存性カリウムチャネルを選択的に阻害します。これによりプルキンエ細胞の活動電位の持続時間が延長し、機能不全の変異型Cav2.1チャネルを通じたカルシウム流入の時間的窓が広がります。結果として、細胞内カルシウム濃度が回復し、不規則になっていたプルキンエ細胞のペースメーカー発火が正常に近い精度へと回復します。EA2の根本病態である「ペースメーカー機能の破綻」を直接修復する、極めて理にかなった薬理学的アプローチです。
EA2患者さんを対象としたプラセボ対照無作為化クロスオーバー試験(RCT)において、ファンプリジン(1日20mg)はアセタゾラミド(1日750mg)と比較しても優れた結果を示しました。
| 評価項目 | ファンプリジン | アセタゾラミド |
|---|---|---|
| プラセボ比 発作減少率 | 63%減少(95%CI 54-74%) | 52%減少 |
| 有害事象発生率 | 26.5%(軽度の感覚異常・疲労) | 44.9%(味覚障害・胃腸障害ほか) |
この結果から、4-AP/ファンプリジンはアセタゾラミドに耐えられない方や効果が不十分な方にとって、極めて有効な代替治療選択肢として位置づけられています。また、アセタゾラミドに耐容性がない場合の代替の炭酸脱水酵素阻害薬として、片頭痛の予防薬としても知られるトピラマートが試みられることもあります。
非薬理学的介入:生活指導とリハビリテーション
🚫 トリガーの特定と回避
極度の身体的ストレス、長時間の激しい運動、過度のカフェイン・アルコール摂取を控える生活指導が基本です。発熱時の早期解熱や十分な睡眠確保も重要です。
🏃 リハビリテーション
進行性の歩行障害や失調に対しては、体幹の安定性強化・筋力維持・バランス感覚改善を目的とした継続的な理学療法が強く推奨されます。
👁 対症療法
複視に対するプリズム眼鏡などの眼科的介入、構音障害に対する言語聴覚士による介入が、必要に応じて行われます。
👶 小児期の包括的支援
神経発達障害やてんかんを合併する小児例では、小児科医・神経内科医・理学療法士・言語聴覚士による多職種連携のもとで、長期的な教育・発達支援を行います。
7. 遺伝カウンセリングの意義
EA2は常染色体顕性遺伝の疾患であるため、確定診断後の家族への丁寧な遺伝カウンセリングが不可欠です。当院での遺伝カウンセリングで扱われる主な内容をご紹介します。
- ➤遺伝形式と再発リスクの説明:常染色体顕性遺伝のため、患者さん本人から子どもへ変異が受け継がれる確率は理論上50%です。ただし、孤発例ではde novo変異であることも多く、両親には変異が認められない場合があります。生殖細胞モザイクの可能性も考慮し、次子の出生前診断についても情報提供を行います。
- ➤予後情報の提供:EA2患者さんの寿命は一般集団と同等であり、疾患自体が直接的な死因となることは通常ありません。一部の患者さんでは若年成人期以降に発作頻度が自然に減少することもあります。一方、長期的には小脳萎縮による慢性失調が進行する可能性も含めて、誠実に情報を共有します。
- ➤出生前診断の選択肢:家族内に既知のCACNA1A変異がある場合、絨毛検査・羊水検査による出生前遺伝子診断が選択肢として存在します。妊娠前のスクリーニングとしては、CACNA1Aを含むインペリアルプランも検討対象となります。
- ➤心理的サポートと生活設計支援:反復する発作によって学業や就労に影響が出ることもあります。職場での合理的配慮の取得、運転制限の検討、家族の理解促進など、生活設計全般を支援します。
8. よくある誤解
誤解①「ただの片頭痛だから様子を見ればよい」
運動誘発性の重度めまいと頭痛は、EA2を強く示唆する組み合わせです。「片頭痛」として放置されているEA2が少なくありません。発作間欠期の眼振と進行性の小脳症状の有無を専門医に評価してもらうことが大切です。
誤解②「家族に同じ症状の人がいないから遺伝病ではない」
EA2は常染色体顕性遺伝ですが、de novo(新生)変異による孤発例も少なくありません。家族歴がないことを理由にEA2を診断から除外することはできません。
誤解③「治療できない遺伝病だから諦めるしかない」
EA2は遺伝性疾患ですが、アセタゾラミドや4-AP/ファンプリジンによる予防治療で発作の頻度・重症度を大幅に減らせる「治療可能な遺伝病」です。早期診断と適切な薬物治療で生活の質を大きく改善できます。
誤解④「内耳のめまいだから耳鼻科だけで診てもらえばいい」
EA2のめまいは中枢性(小脳由来)であり、メニエール病やBPPVなどの内耳疾患とは病態が異なります。神経内科医・臨床遺伝専門医による評価が必要です。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 反復発作性失調症・遺伝性めまい疾患のご相談
EA2をはじめとする遺伝性神経疾患の診断・遺伝カウンセリングは、
臨床遺伝専門医が在籍するミネルバクリニックにご相談ください。
関連記事
参考文献
- [1] Jen JC, Graves TD, Hess EJ, et al. Episodic ataxia type 2. PubMed. PMID:17395137. [PubMed]
- [2] OMIM #108500. Episodic Ataxia Type 2 (EA2). Johns Hopkins University. [OMIM]
- [3] MedlinePlus Genetics. Episodic ataxia. National Library of Medicine. [MedlinePlus]
- [4] MedlinePlus Genetics. CACNA1A gene. National Library of Medicine. [MedlinePlus]
- [5] Bird TD. Hereditary Ataxia Overview. GeneReviews®. University of Washington, Seattle. [GeneReviews / NCBI Bookshelf]
- [6] Choi KD, Choi JH. Episodic Ataxias: Clinical and Genetic Features. J Mov Disord. 2016. [J Mov Disord]
- [7] Indelicato E, Boesch S. From Genotype to Phenotype: Expanding the Clinical Spectrum of CACNA1A Variants in the Era of Next Generation Sequencing. Front Neurol. 2021. [Frontiers in Neurology]
- [8] Clinical and genetic characterization of CACNA1A-related disease. PMC9458680. [PMC9458680]
- [9] Strupp M, Kalla R, Claassen J, et al. A Randomized Trial of 4-Aminopyridine in EA2 and Related Familial Episodic Ataxias. Neurology / PMC3136055. [PMC3136055]
- [10] Muth C, Strupp M, et al. Fampridine and Acetazolamide in EA2 and Related Familial EA: A Prospective Randomized Placebo-Controlled Trial. PMC8382428. [PMC8382428]
- [11] Etzion Y, Grossman Y. The Therapeutic Mode of Action of 4-Aminopyridine in Cerebellar Ataxia. J Neurosci / PMC2909847. [PMC2909847]
- [12] Diagnostic approach to episodic ataxia types 1 and 2: a proposed algorithm. Front Neurol. [Frontiers in Neurology]
- [13] Orphanet. Episodic ataxia type 2. ORPHA:97. [Orphanet]






