目次

CACNA1A遺伝子は、脳の神経細胞でカルシウムの出入りを制御する「P/Q型カルシウムチャネル」を作るための、神経活動の根幹を担う遺伝子です。この一つの遺伝子に変異が生じると、片麻痺を伴う片頭痛・周期的な小脳失調・進行性の小脳変性症・重度の発達性てんかん性脳症まで、変異のタイプによって全く異なる神経疾患を引き起こすという、極めてユニークな特徴を持っています。
Q. CACNA1A遺伝子とは何ですか?まず結論だけ知りたいです
A. CACNA1A遺伝子は、脳のカルシウムチャネル(CaV2.1)の主要部品を作る遺伝子で、19番染色体に位置しています。変異のタイプ次第で、家族性片麻痺性片頭痛1型(FHM1)・反復発作性失調症2型(EA2)・脊髄小脳変性症6型(SCA6)・発達性てんかん性脳症などの複数の神経疾患を引き起こすことが知られており、近年は知的障害や自閉症スペクトラムを含む幅広い表現型が認識されています。
- ➤遺伝子の場所と機能 → 19p13.13、P/Q型カルシウムチャネル(CaV2.1)α1Aサブユニットをコード
- ➤特殊な構造 → 一つの遺伝子から2つの異なるタンパク質を作る「バイシストロニック構造」
- ➤変異タイプ → 機能獲得型(GoF)・機能喪失型(LoF)・CAGリピート伸長の3パターン
- ➤関連疾患 → FHM1・EA2・SCA6・DEE42を含む幅広い神経発達障害スペクトラム
- ➤最新治療 → アンチセンス核酸(ASO)によるSCA6標的療法が臨床応用へ前進中
1. CACNA1A遺伝子とは:基本情報と神経活動における役割
CACNA1A遺伝子は、ヒトの19番染色体短腕(19p13.13)に位置する、神経科学のなかでも特に重要な遺伝子の一つです。この遺伝子の役割を一言で表すと、「脳の神経細胞が情報をやり取りするための、いわば『門』を作る設計図」です。具体的には、神経細胞の膜上に存在するP/Q型電位依存性カルシウムチャネル(CaV2.1)の主要部品であるα1A(アルファ・ワン・エー)サブユニットというタンパク質をコードしています。
💡 用語解説:電位依存性カルシウムチャネルとは
神経細胞の膜には、特定の物質を通すための小さな「門」がたくさん存在します。電位依存性カルシウムチャネルは、細胞膜の電気的な状態が変化すると開き、カルシウムイオン(Ca²⁺)を細胞の外から中へ流入させる専用の通路です。このカルシウムの流入が、神経細胞が次の神経細胞へ情報を伝える「神経伝達物質の放出」の引き金になります。チャネルがうまく働かないと、脳の情報伝達がスムーズにいきません。
CaV2.1チャネルは中枢神経系全体に広く分布していますが、特に運動の協調を司る小脳のプルキンエ細胞と顆粒細胞に大量に発現しています。これがCACNA1A遺伝子の変異で小脳症状(ふらつき・運動失調)が高頻度に出現する解剖学的な理由です。さらに、神経細胞の生存維持・シナプス可塑性(記憶や学習の基盤)にも深く関わっています。
💡 用語解説:プルキンエ細胞とは
小脳皮質に存在する、非常に大きく枝分かれした樹状突起を持つ神経細胞です。小脳の出力を一手に担う「司令塔」のような存在で、運動の精密なコントロール・タイミング調整・運動学習に欠かせません。CACNA1A遺伝子に変異があると、このプルキンエ細胞の働きが乱れ、ふらつきや構音障害(しゃべりにくさ)が出現します。
2. 構造の特異性:一つの遺伝子が二つのタンパク質を作る
CACNA1A遺伝子の最大の特徴であり、世界中の神経遺伝学者を魅了している点が、「バイシストロニック構造」と呼ばれる極めて珍しい仕組みです。通常、一つの遺伝子は一つのタンパク質を作るのが原則ですが、CACNA1Aは全く役割の異なる2つのタンパク質を同じ遺伝子から生み出します。
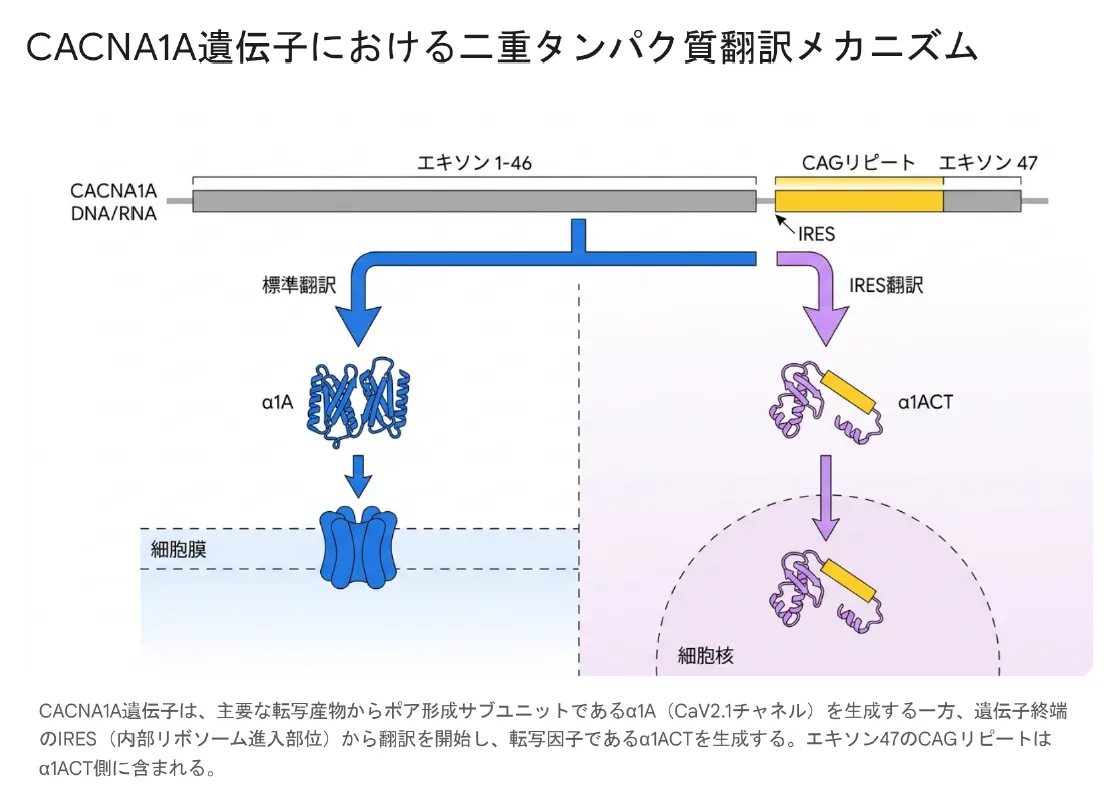
CACNA1A遺伝子は主要転写物からα1A(CaV2.1チャネル)を生成する一方、遺伝子終端のIRES(内部リボソーム進入部位)から翻訳を開始し、転写因子α1ACTを生成する。エキソン47のCAGリピートはα1ACT側に含まれる。
タンパク質①:α1A(カルシウムチャネル本体)
遺伝子の主要部分から翻訳されるα1Aサブユニットは、4つの巨大な膜貫通リピート(I〜IV)を持ち、それぞれが6つのαヘリックス(S1〜S6)で構成されています。S4ヘリックスは電圧センサー、S5・S6ヘリックスはカルシウムが実際に通過する「イオン伝導孔(ポア)」を裏打ちする、機能的に最も重要な領域です。
タンパク質②:α1ACT(核内転写因子)
遺伝子の終端付近にあるIRES(内部リボソーム進入部位)と呼ばれる隠れた翻訳開始点から、もう一つのタンパク質「α1ACT」が独立して作られます。α1ACTはイオンチャネルではなく、細胞核に移行して他の遺伝子の働きを制御する転写因子として機能し、特に小脳プルキンエ細胞の正常な発達・成長に欠かせません。
💡 用語解説:IRES(内部リボソーム進入部位)とは
通常、リボソーム(タンパク質を作る分子機械)はメッセンジャーRNAの「先頭」から翻訳を始めます。しかしIRESがあると、リボソームはRNAの途中から翻訳を開始できます。CACNA1A遺伝子はこの仕組みを使って、一つのRNAから2種類のタンパク質を作り分けているのです。この発見は、長い間「ジャンク」と思われていた遺伝子領域に実は重要な機能が隠れていることを示した画期的な研究でした。
エキソン47のCAGリピート:もう一つの重要な特徴
遺伝子の尾部、α1ACTがコードされる最終エキソン(エキソン47)の内部には、CAG(シトシン-アデニン-グアニン)という3つのDNA塩基配列が繰り返される「トリヌクレオチドリピート」が存在します。健康な人では4〜18回の繰り返しに収まっていますが、これが20〜33回まで異常に伸びると、脊髄小脳変性症6型(SCA6)を発症します。
3. 関連する4つの主要な神経疾患
CACNA1A遺伝子は歴史的に3つの古典的な家族性神経疾患の原因として知られていましたが、近年は次世代シーケンス技術の普及により、4つ目の主要表現型として「発達性てんかん性脳症」が加わりました。それぞれの疾患は、変異のタイプによって特徴的な臨床像を示します。
4. 変異のタイプと病態メカニズム
CACNA1A遺伝子の変異は、チャネルの働きに対して「機能獲得型」「機能喪失型」「リピート伸長型」の3つの全く異なる影響を与えます。この違いが、引き起こされる疾患の決定的な差を生みます。
💡 用語解説:ミスセンス変異とde novo(新生)変異
ミスセンス変異とは、DNA塩基が1つ変わることで、タンパク質を構成するアミノ酸が別の種類に置き換わる変異のこと。タンパク質の形や機能に影響を与えます。
de novo(新生)変異とは、両親には存在せず、お子さんで初めて生じた変異のことです。CACNA1A関連の重症例(DEE42など)の多くはde novo変異によって発症します。
機能獲得型(GoF)変異:チャネルが「開きすぎる」
機能獲得型変異では、カルシウムチャネルが通常より低い刺激で開きやすくなったり、一度開くと閉じるのに時間がかかるといった変化が生じます。その結果、神経細胞内へのカルシウム流入が異常に増え、神経細胞が過剰興奮状態に陥ります。FHM1や難治性てんかん重積状態と強く関連し、皮質拡延性抑制(脳の異常な電気的波及)の原動力になります。
⚠️ 特に注意すべきS218L変異
S218L変異を持つ小児では、スポーツ中の軽い接触や日常の些細な頭部打撲がきっかけで、致死的な脳浮腫や長期昏睡が引き起こされるリスクがあります。この変異が同定された場合は、頭部に衝撃を受けるリスクのある活動について、保護者と医療チームで慎重な話し合いが必要です。
機能喪失型(LoF)変異:チャネルが「開きにくくなる」
機能喪失型変異では、チャネルの立体構造の異常や細胞膜への配置の失敗により、カルシウム流入が病的に減少します。神経伝達物質の放出が滞り、神経回路全体のシグナル伝達効率が落ちます。EA2や欠神発作を伴うてんかんと強く関連します。
CAGリピート伸長:毒性タンパク質の蓄積(SCA6)
SCA6は前述の2タイプとは全く異なる病態メカニズムを持ちます。エキソン47のCAGリピートが伸長すると、転写因子α1ACTのカルボキシル末端に異常に長いポリグルタミン鎖が形成され、細胞内に毒性の凝集体として蓄積。これが特に小脳プルキンエ細胞を選択的に変性・死滅させます。ハンチントン病と同じ「ポリグルタミン病」のカテゴリーに含まれます。
SCA6のCAGリピート数判定基準とハンチントン病との比較
SCA6のCAGリピート伸長は、同じポリグルタミン病であるハンチントン病(HD)と比較して、驚くほど少ないリピート数で発症するという臨床上極めて重要な特徴があります。発症前診断や遺伝カウンセリングでは、以下の判定基準が国際的な標準として用いられています。
| 判定区分 | SCA6(CACNA1A) | ハンチントン病(HD)参考 |
|---|---|---|
| 正常アレル | 18回以下 | 26回以下 |
| 中間アレル (次世代で伸長リスク) |
SCA6では極めて稀 | 27〜35回 |
| 不完全浸透アレル (発症しない場合あり) |
19〜20回 | 36〜39回 |
| 完全浸透アレル (生涯のどこかで発症) |
21〜33回 | 40回以上 |
この表が示す事実は、患者さんとご家族にとって非常に大きな意味を持ちます。SCA6では「21回」というハンチントン病なら完全に「正常」と判定される短いリピート数で、完全浸透(生涯のどこかで発症する可能性が極めて高い状態)となります。ただし、完全浸透アレルでもリピート数が短い場合(21〜23回など)は発症年齢が遅くなる(50〜60代以降)傾向があり、リピート数が増えるほど若年化します。
なぜわずかなリピート伸長で発症するのか
ハンチントン病など多くのポリグルタミン病が36〜40回以上の長い伸長で発症するのに対し、SCA6はわずか19〜33回という短い伸長で発症します。この特異な現象には、SCA6独自のメカニズムがあります。
🔬 SCA6の発症閾値が低い2つの理由
① タンパク質サイズとの比率:異常の舞台となるα1ACTは、分子量約75kDaと比較的小さなタンパク質断片です。ハンチンチンのような巨大タンパク質の一部が伸びる疾患とは異なり、小さなα1ACTでは数個のグルタミン追加でも全体の立体構造を決定的に破壊してしまいます。
② ダイレクトな核内毒性:α1ACTはプルキンエ細胞の核に移動し、直接DNAに結合して神経発生を制御する精密な転写因子です。わずかな構造変化で本来の標的(GRN遺伝子など)に結合できなくなり、核内ですぐに有毒な凝集体を形成して直接的に細胞死を引き起こします。
なぜプルキンエ細胞だけが選択的に変性するのか
CACNA1Aの変異は全身の細胞に存在するにもかかわらず、SCA6では小脳のプルキンエ細胞だけが選択的にダメージを受けます(選択的脆弱性)。この理由もα1ACTの特異な働きにあります。
- ➤小脳での圧倒的なα1ACT産生量:α1ACTを作り出すIRES(内部リボソーム進入部位)の活性は、ヒトの体の中でも小脳で極めて高いことが分かっています。そのため毒性を持つ変異型α1ACTが小脳に大量に蓄積します。
- ➤生存ネットワークの要としてのα1ACT:α1ACTは転写因子として、神経細胞の生存や成長に不可欠な遺伝子群(GRN、PMCA2など)を直接コントロールしています。複雑で大きな樹状突起を持つプルキンエ細胞はこれらの遺伝子への依存度が他細胞より圧倒的に高いため、α1ACTの異常が直ちに細胞死につながります。
この「変異が起きるタンパク質の特殊性」と「小脳での発現量の高さ」が掛け合わさることで、SCA6の低い発症閾値とプルキンエ細胞への選択的毒性という独特の病態が生まれます。これは前述したIRES標的ASO療法が「α1Aチャネルには影響を与えず、毒性α1ACTのみを選択的に止める」という戦略を理論的に支える重要な根拠でもあります。
CACNA1A関連障害の症状発現頻度(47名コホート研究)
2022年に発表された47名の自然歴コホート研究では、CACNA1A病原性変異を持つ小児期発症患者の症状頻度が定量的に明らかにされました。以下のグラフが示すように、想像以上に重篤な神経発達障害が高頻度で見られます。
📊 CACNA1A関連障害における主要症状の発現頻度(n=47)
出典:Indelicatoら(2022), PMC9458680。てんかんを合併する患者の69%という高い割合でてんかん重積状態が発生する。
タンパク質構造と臨床重症度の精密な対応
最近の構造生物学的研究により、変異がα1Aサブユニットの「どの位置」に生じたかが、臨床重症度と密接に対応することが明らかになっています。イオンが直接通過するS5・S6ヘリックス内の変異を持つ患者群は、S3ヘリックスやリンカー領域に変異を持つ患者群と比べて、有意に重症度が高くなります。たとえばp.Val1392Met(ポアドメイン)はてんかんおよびてんかん重積状態の頻度が著しく高いことが報告されています。
5. 診断と遺伝子検査の進め方
CACNA1A関連疾患の診断は、表現型のオーバーラップが大きいため、従来の単一遺伝子検査では見逃されることが少なくありません。現在は次世代シーケンス(NGS)を用いた網羅的アプローチが世界標準となっています。
多層的な遺伝子検査戦略
- ➤全エクソームシーケンス(WES)または遺伝子パネル:原因不明の発達性てんかん性脳症や非典型的な失調症の第一選択。点変異・小欠失・小挿入を効率的に検出します。
- ➤CAGリピート伸長解析(PCR法):標準的なNGSではSCA6のCAGリピート伸長を正確に検出できません。成人発症の進行性失調症が疑われる場合は、PCRベースの専用解析が必須です。
- ➤家族解析(トリオ解析):両親も含めた3名で同時解析することで、de novo変異の同定精度が大幅に向上します。
反復発作性失調症の鑑別診断
エピソード性の運動失調を呈する患者では、カリウムチャネル遺伝子KCNA1の変異による反復発作性失調症1型(EA1)との鑑別が最も重要です。両者は治療反応性も異なるため、正確な診断が必要です。
| 鑑別項目 | EA1(反復発作性失調症1型) | EA2(反復発作性失調症2型) |
|---|---|---|
| 原因遺伝子 | KCNA1(カリウムチャネル) | CACNA1A(カルシウムチャネル) |
| 発作の持続時間 | 数秒〜数分(短時間) | 数分〜数日間(長時間) |
| 発作間欠期の所見 | ミオキミア(筋のさざ波) | 下向き眼振、注視誘発眼振 |
| 合併症 | 筋けいれん、こわばり | 片頭痛(約50%)、めまい |
| MRI所見 | 通常は正常 | 小脳虫部の萎縮 |
| 治療反応性 | カルバマゼピン | アセタゾラミド、4-アミノピリジン |
6. 治療と最新研究動向
CACNA1A関連疾患の根本治療はまだ存在しませんが、変異タイプに応じた薬物療法と、遺伝子レベルでの根治を目指す次世代治療の開発が急速に進んでいます。
現行の薬物療法
💊 アセタゾラミド
EA2の第一選択薬として長年使用される炭酸脱水酵素阻害薬。患者の49〜70%で発作頻度・重症度の顕著な改善が報告されています。脳組織の局所pH低下を介して興奮性NMDA受容体を抑制し、抑制性GABAa受容体を増強する複合作用が解明されつつあります。
💊 4-アミノピリジン/ファンプリジン
アセタゾラミドに反応しない場合の第二選択。EAT2TREAT試験ではプラセボと比べて発作回数を63%に減少。極めて低濃度でKv1.5カリウムチャネルを選択的に遮断し、乱れていたプルキンエ細胞のペースメーキング機能を復元します。
💊 ベラパミル(GoF変異向け)
機能獲得型変異のFHM1患者に対する精密医療の試み。カルシウム拮抗薬として理論上、過剰なカルシウム流入を直接ブロック。小規模報告で肯定的な反応が見られ、今後のプラセボ対照試験が期待されています。
次世代治療:アンチセンス核酸(ASO)によるSCA6標的治療
SCA6の根本治療として世界中の研究者が最も注目しているのが、アンチセンス核酸(ASO)やマイクロRNAを用いた「特異的翻訳阻害技術」です。
💡 用語解説:アンチセンスオリゴヌクレオチド(ASO)
病気の原因となる特定のRNAだけにくっついて、そのRNAからタンパク質が作られるのを止める、20塩基程度の短いDNAやRNAの断片です。すでに脊髄性筋萎縮症(SMA)の治療薬「ヌシネルセン」などで実用化されており、希少な神経疾患の「狙い撃ち治療」を実現する技術として急速に発展しています。
SCA6治療の難しい点は、原因遺伝子のCACNA1A全体を止めてしまうと、生命維持に必須なα1Aチャネルまで失われて致死的になってしまうこと。研究チームはこの問題を解決するため、IRES配列に結合してリボソームをブロックするASOやmiRNA-3191-5pを開発しました。これにより、必須なα1Aチャネルの正常な生成は維持しつつ、毒性α1ACTの翻訳のみを選択的に止めることが可能になりました。重症マウスモデルでの前臨床試験では運動失調の進行が劇的に停止することが証明されており、成人SCA6患者への臨床試験が最も期待される治療戦略です。
発達性てんかん性脳症(DEE)への新薬開発
CACNA1A変異を含む難治性のDEE患者を対象に、複数の新薬の国際共同臨床試験が進行中です。ベキシカセリン(次世代の選択的セロトニン受容体アゴニスト)はDEEpOCEAN試験として17カ国80施設で第3相試験中、レルトリジン(ナトリウムチャネル調節薬)はEmerald試験として有効性を評価中です。
7. 遺伝カウンセリングのポイント
CACNA1A関連疾患の確定診断後は、ご家族への丁寧な遺伝カウンセリングが欠かせません。情報提供は中立的に行い、最終的な意思決定はご家族に委ねるスタンスを大切にしています。
💡 用語解説:常染色体顕性遺伝(旧名:常染色体優性遺伝)
2本ある常染色体(性染色体以外の染色体)のうち、片方の遺伝子に変異があれば症状が現れる遺伝形式です。CACNA1A関連疾患はこの遺伝形式をとり、患者さんが子どもを持つ場合、お子さんが変異を受け継ぐ確率は理論上50%です。ただし発達性てんかん性脳症など重症型の多くはde novo(新生)変異によるもので、両親には変異が見つからないケースが大半です。
- ➤遺伝形式と再発リスクの説明:変異タイプ・家系内発症パターンを丁寧に整理し、ご家族個別の状況に合わせて説明します。
- ➤予後と長期管理計画:変異タイプによって予後が大きく異なるため、可能性のある経過と必要な医療体制を共有します。
- ➤出生前診断の選択肢:既知の変異が家族内にある場合は、絨毛検査・羊水検査による確定診断が可能です。次子を望むご家族にはインペリアルプランなどのスクリーニング選択肢もあります。
- ➤S218L変異など特殊変異への配慮:頭部外傷リスクなど生活管理上の注意点を保護者と医療チームで共有します。
8. よくある誤解
誤解①「片頭痛の遺伝子=大きな問題ではない」
FHM1のイメージが強いCACNA1Aですが、変異タイプによっては重度のてんかん重積状態・致死的な脳浮腫・知的障害を伴う重篤な疾患を引き起こします。「片頭痛だから軽症」という認識は危険です。
誤解②「親に症状がないから遺伝ではない」
CACNA1A関連の重症型(DEE42など)の多くはde novo(新生)変異。ご両親には症状も変異もなく、お子さんで初めて生じた変異であることが大半です。「家族に同じ病気がいないから遺伝病ではない」とは限りません。
誤解③「NGSをやればSCA6もわかる」
標準的な次世代シーケンスでは、CAGリピートの伸長を正確に検出できません。成人発症の進行性失調症が疑われる場合は、必ずPCRベースのリピート伸長解析を並行して行う必要があります。
誤解④「同じ変異なら同じ症状が出る」
CACNA1Aは浸透率や表現型の幅が大きい遺伝子です。家系内でも片頭痛だけの方、ふらつきが目立つ方、てんかんが前面に出る方など、症状の表れ方が異なるケースが多数報告されています。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 CACNA1A関連疾患・神経遺伝病のご相談
CACNA1A遺伝子に関わる神経疾患の診断・治療方針・出生前診断について、
臨床遺伝専門医にお気軽にご相談ください。
関連記事
参考文献
- [1] MedlinePlus Genetics. CACNA1A gene. National Library of Medicine. [MedlinePlus]
- [2] Indelicato E, et al. Clinical and genetic characterization of CACNA1A-related disease. Clin Genet. 2022. [PMC9458680]
- [3] Jen JC, et al. Familial Hemiplegic Migraine. GeneReviews® [Internet]. [GeneReviews NBK1388]
- [4] Spinocerebellar Ataxia Type 6. GeneReviews® [Internet]. [GeneReviews NBK1140]
- [5] Mantuano E, et al. Mutation Spectrum in the CACNA1A Gene in 49 Patients with Episodic Ataxia. Sci Rep. 2017. [PMC5451382]
- [6] Du X, et al. Targeting the CACNA1A IRES as a treatment for spinocerebellar ataxia type 6. Cerebellum. 2018. [PMC5809202]
- [7] Alviña K, Khodakhah K. The Therapeutic Mode of Action of 4-Aminopyridine in Cerebellar Ataxia. J Neurosci. 2010. [PMC2909847]
- [8] Strupp M, et al. Fampridine and Acetazolamide in EA2 and Related Familial EA: A Prospective Randomized Placebo-Controlled Trial. Neurol Genet. 2021. [PMC8382428]
- [9] CACNA1A Foundation. Clinical Trials & Research. [CACNA1A Foundation]
- [10] OMIM #601011. CACNA1A Gene. Johns Hopkins University. [OMIM]






