目次

家族性片麻痺性片頭痛1型(FHM1)は、19番染色体上のCACNA1A遺伝子の変異によって発症する、人口およそ1万人に1人未満という極めて稀な遺伝性の片頭痛です。一般的な片頭痛と決定的に違うのは、発作のたびに体の片側に運動麻痺(片麻痺)が起こり、それが数時間から数日続くこと。さらに患者さんのおよそ40〜50%が加齢とともに進行する小脳失調症も合併します。
Q. FHM1ってどんな病気ですか?普通の片頭痛と何が違うの?
A. CACNA1A遺伝子の変異が原因で起こる、運動麻痺を伴う特殊な片頭痛です。発作時に体の片側がしびれて動かなくなる「片麻痺」が数時間〜数日続き、患者さんの40〜50%が進行性の小脳失調症を合併します。一般的な片頭痛薬(トリプタン製剤)が禁忌のため、遺伝子検査による正確な診断が極めて重要です。
- ➤疾患の定義 → OMIM #141500、ICHD-3コード1.2.3.1.1、有病率1万人に1人未満
- ➤原因と機序 → CACNA1A遺伝子変異によるCaV2.1チャネル機能亢進と皮質拡延性抑制
- ➤主な症状 → 運動麻痺を伴う前兆、高熱や昏睡を伴う重症発作、進行性小脳失調
- ➤代表的な変異 → T666M(最頻)、R583Q、S218L(外傷後昏睡リスク)
- ➤治療 → トリプタン禁忌、アセタゾラミド・4-アミノピリジンによる失調管理
1. 家族性片麻痺性片頭痛1型(FHM1)とは:疾患の定義と疫学
家族性片麻痺性片頭痛(Familial Hemiplegic Migraine: FHM)は、片頭痛のなかでも特に稀な「前兆を伴う片頭痛」の亜型です。最大の特徴は、発作のたびに体の片側に運動麻痺(片麻痺)が起こり、これが数時間から数日続くこと。第一度または第二度近親者(親・きょうだい・祖父母など)に同じ症状の方がいる場合に「家族性(FHM)」と診断され、家族歴がない孤発例は「孤発性片麻痺性片頭痛(SHM)」として分類されます。
この疾患は、原因となる遺伝子によって3つのタイプに分かれます。19番染色体短腕(19p13)にあるCACNA1A遺伝子の変異によって起こるものを「家族性片麻痺性片頭痛1型(FHM1)」と呼びます。FHM1はFHM全体のおよそ50〜70%を占める最も頻度の高いタイプであり、単なる発作性の頭痛にとどまらず、慢性的に進行する小脳の障害を高頻度に合併することで知られています。
💡 用語解説:常染色体顕性(優性)遺伝
FHM1は「常染色体顕性遺伝(じょうせんしょくたいけんせいいでん)」、かつては「常染色体優性遺伝」と呼ばれていた遺伝形式の疾患です。「常染色体」とは性染色体(X・Y)以外の染色体のことで、「顕性(優性)」とは、ペアで存在する2本の染色体のうちどちらか1本に変異があるだけで症状が現れることを意味します。親が患者さんの場合、お子さんに50%の確率で遺伝します。男女どちらにも同じ確率で発症します。
疫学:希少だが世界中で報告される疾患
片麻痺性片頭痛全体の有病率は人口およそ1万人に1人(0.01%)と推定されており、FHMとSHMの頻度はほぼ同等です。1999年にデンマークで520万人を対象に行われた大規模な疫学調査では、FHMの有病率は0.005%と算出されました。1910年に最初の症例が報告されて以来、世界中で詳細に記述された家系は約150家系にとどまり、極めて希少な疾患であることが裏付けられています。
発症年齢は一般的な片頭痛よりも早く、多くは小児期から思春期(10歳〜20歳まで)に最初の発作を経験します。一般的な片頭痛は女性が男性の2〜3倍多いという顕著な性差がありますが、FHM1ではこの性差はやや弱まり、男女ともに発症します。
2. 原因遺伝子CACNA1AとCaV2.1チャネル機能亢進
FHM1の原因は、19番染色体短腕(19p13)にあるCACNA1A遺伝子の変異です。この遺伝子は、神経細胞の表面に存在する「P/Q型電位依存性カルシウムチャネル(CaV2.1チャネル)」というタンパク質の主要部分(α1サブユニット)の設計図を持っています。
💡 用語解説:CaV2.1チャネル(P/Q型カルシウムチャネル)
脳の神経細胞は、電気信号と化学物質(神経伝達物質)を組み合わせて情報をやりとりしています。CaV2.1チャネルは、神経細胞の末端に並んでいる「カルシウムイオンの出入り口」のひとつ。電気信号が来るとパッと開いて、外から細胞の中にカルシウムを流し込みます。このカルシウムがスイッチとなって、グルタミン酸という興奮性の神経伝達物質が放出され、隣の神経細胞へ信号が伝わる仕組みです。特に大脳皮質や小脳のプルキンエ細胞で活発に働いています。
💡 用語解説:機能亢進(Gain-of-function)
遺伝子変異の影響は大きく2種類に分かれます。「機能喪失(Loss-of-function)」はタンパク質の働きが減るパターン。「機能亢進(Gain-of-function)」は逆に、タンパク質が働きすぎてしまうパターンです。FHM1のCACNA1A変異は機能亢進型で、CaV2.1チャネルが本来より低い電圧で開きやすくなり、あるいは開いている時間が長くなることで、細胞内へのカルシウム流入が過剰になります。
シナプスでの異常な情報伝達——グルタミン酸の暴走
FHM1を引き起こすミスセンス変異は、チャネルのポア(穴)の内壁や電位センサー領域といった機能的に重要な部位に集中しています。これらの変異によってCaV2.1チャネルがいわば「過剰反応モード」になり、神経細胞のシナプス前末端に大量のカルシウムが流入します。その結果、興奮性神経伝達物質グルタミン酸の放出が異常に増大し、大脳皮質全体が過興奮状態に陥ります。
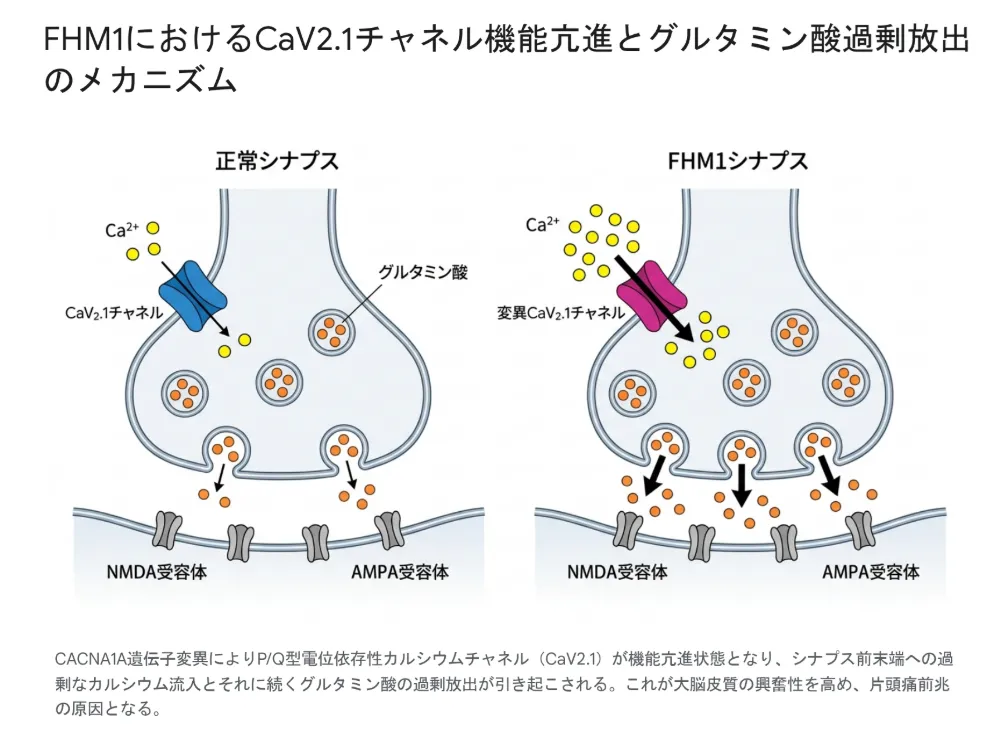
正常シナプス(左)では適切な量のカルシウム流入とグルタミン酸放出が起こる。一方FHM1シナプス(右)では変異CaV2.1チャネルを介して過剰なカルシウムが流入し、グルタミン酸が大量に放出される結果、NMDA・AMPA受容体が過剰に刺激されて大脳皮質の興奮性が異常に高まる。
皮質拡延性抑制(CSD):前兆症状の電気生理学的正体
💡 用語解説:皮質拡延性抑制(CSD)
Cortical Spreading Depression(CSD)と呼ばれる現象で、大脳皮質の表面を秒速数ミリメートルでゆっくりと広がっていく強烈な脱分極の波のこと。神経細胞が一斉に興奮した直後にいったん機能停止することで、その領域の皮質機能が一時的にストップします。波が視覚野を通れば視野欠損や閃輝暗点(チカチカした光)として、運動野を通れば片麻痺として、感覚野を通ればしびれとして自覚されます。これが片頭痛の「前兆(オーラ)」の正体です。
グルタミン酸が大量にあふれると、隣接する神経細胞が次々に過興奮を起こし、CSDの発生閾値が極端に下がります。FHM1ではこのCSDが起こりやすく、しかも強力に伝播するため、一般的な片頭痛でみられる視覚性前兆だけにとどまらず、運動野の機能を停止させて重篤な片麻痺を引き起こし、さらに脳幹の網様体を巻き込むことで意識障害にまで進展することがあると考えられています。
FHMの3つのタイプ:原因遺伝子による分類
| サブタイプ | 原因遺伝子 | 染色体 | 機能変化 | 特徴 |
|---|---|---|---|---|
| FHM1 | CACNA1A | 19p13 | 機能亢進 | FHM全体の50〜70%。進行性小脳失調を高頻度に合併 |
| FHM2 | ATP1A2 | 1q23 | 機能喪失 | 重篤な神経症状はあるが、小脳症状の合併は明確ではない |
| FHM3 | SCN1A | 2q24 | 機能亢進 | 極めて稀。てんかん性疾患とのオーバーラップが強い |
3. 主な症状:発作のプロファイルと重症化
典型的なFHM1の発作は、5分から20分かけて徐々に進展する前兆(オーラ)から始まります。一般的な片頭痛の前兆が60分以内に消失するのに対して、FHM1の運動麻痺は数時間から数日続くことが特徴です。頭痛そのものが治まったあとも、片麻痺だけが長く残ってしまうケースが珍しくありません。
🌀 前兆症状(オーラ)
- 運動麻痺(片側の手足・顔の脱力)
- 視覚症状(閃輝暗点・暗点)
- 感覚症状(チクチク感・しびれ)
- 言語障害(失語・構音障害)
- 脳底型症状(めまい・耳鳴り・複視)が最大70%
⏰ 頭痛と持続時間
- 拍動性の片側頭痛
- 4時間〜72時間持続
- 前兆直後、または1時間以内に発現
- 頭痛を伴わない「無頭痛性片頭痛」の場合も
- 発作頻度は平均年3回程度
🚨 重症発作
- 長期にわたる片麻痺(数日〜数週)
- 38度を超える高熱
- 急性脳症・錯乱状態
- 昏睡
- 全身性てんかん発作
🔍 髄液検査の特徴
- 急性期に初圧の著明な上昇
- 細胞増多(単核球優位)
- ウイルス性・細菌性髄膜脳炎との類似
- 急性脳卒中・感染症との鑑別が困難
⚠️ 臨床的に最も注意すべきポイント:FHM1の重症発作は、長時間の片麻痺・高熱・意識障害・てんかんを伴うため、初期診療の現場では急性脳卒中・髄膜脳炎・てんかん重積状態と誤診されやすいのです。脳脊髄液検査でも炎症性変化を示すため鑑別はさらに困難になります。家族歴の確認と遺伝子検査の重要性がここにあります。

4. 進行性小脳失調症の合併——FHM1を特別な疾患にする決定的特徴
FHM1を他の片麻痺性片頭痛(FHM2・FHM3・SHMの一部)と決定的に区別する特徴が、慢性かつ進行性の小脳症状の合併です。FHM全体では非発作時に永続的な小脳症状を持つ家系は約20%ですが、CACNA1A変異が確認されたFHM1家系に限ると、その割合は40〜50%へと跳ね上がります。
💡 用語解説:小脳失調症と眼振
小脳は脳の後ろ下方にある「運動の調整役」。動作のなめらかさ・タイミング・バランスを担っています。小脳が障害されると、歩行のふらつき(体幹失調)、目的物に手を伸ばすとずれてしまう(測定異常)、目標近くで震える(企図振戦)、ろれつが回らない(構音障害)などが現れます。眼振(がんしん)は眼球が意思と無関係に揺れ動く現象で、水平方向・下向きなどパターンがあり、小脳症状の早期サインとして重要です。
二相性の臨床経過——若年期は頭痛、中年期以降は失調が前景に
小脳症状の経過は年齢に大きく依存します。多くの場合、10代から20代の若年期には重篤な片麻痺発作が前景に立ち、小脳症状は発作時の一過性のものか、非発作時の軽度な眼振にとどまります。しかし、加齢とともに小脳変性がじわじわと進行し、中年期以降には歩行障害や構音障害が永続的なものとして定着します。
興味深いことに、年齢を重ねるほど頭痛発作の頻度は徐々に減少する一方で、小脳症状の重症度は増していくという二相性の経過をたどる患者さんが多いのが特徴です。MRIでは小脳虫部上部の萎縮が顕著に観察され、進行例では小脳半球全体の容量低下に及びます。
小児期から現れる多面的な神経症状
CACNA1A遺伝子は中枢神経系全般で広く機能しているため、片麻痺と小脳失調以外にもさまざまな症状を重ねることがあります。てんかんの合併リスクが高く、小児期に欠神てんかんが初発症状となるケースや、重度では発達性てんかん性脳症42型として現れる例もあります。また乳幼児期からの運動発達遅延、後天的な知的障害、小児期特有の発作性上方注視といった眼球運動異常が、他の神経症状に先行する初期兆候として現れることが知られています。
5. ICHD-3に基づく診断基準
FHM1の診断は、国際頭痛学会(IHS)が定める国際頭痛分類第3版(ICHD-3)の厳格な基準(コード1.2.3.1.1)に基づきます。確定診断には、特異的な臨床症状の病歴と家族歴、そしてCACNA1A遺伝子の病原性変異の同定の3つが揃う必要があります。
| 診断項目 | 基準の詳細 |
|---|---|
| A. 発作回数 | 基準BおよびCを満たす発作が、生涯に少なくとも2回以上 |
| B. 前兆の性質 | 完全可逆性の運動麻痺を伴う前兆+以下のうち1つ以上:①視覚症状、②感覚症状、③言語・発話障害、④脳幹症状、⑤網膜症状 |
| C. 前兆の進展 | ①前兆症状が5分以上かけて徐々に進展、②2つ以上の前兆が連続、③各前兆の持続時間が5〜60分(運動麻痺は72時間以上でも可)、④少なくとも1つは片側性、⑤少なくとも1つは陽性症状、⑥前兆中または前兆後60分以内に頭痛発現——のうち3つ以上 |
| D. 家族歴 | 第一度または第二度近親者の1人が「運動麻痺を伴う前兆」を有する片頭痛患者 |
| E. 遺伝子変異 | 遺伝子検査でCACNA1A遺伝子に病原性変異が同定されること |
6. 遺伝子型と表現型相関——変異ごとに異なる臨床像
FHM1の症状の深刻さや複雑さは、CACNA1A遺伝子のどの位置に変異が起きたかによって大きく異なります。これまでに少なくとも15種類のミスセンス変異が同定され、複雑な遺伝子型-表現型相関を形成しています。なかでも創始者効果(同一の祖先に由来すること)なしに複数の家系で繰り返し報告される「再発性変異」として、T666M、R583Q、S218L、R1668W、I1811Lなどが知られています。
💡 用語解説:ミスセンス変異と新生突然変異
ミスセンス変異とは、DNAの塩基がひとつ変わることで、タンパク質を構成するアミノ酸が別の種類に置き換わるタイプの変異です。タンパク質の形がわずかに変わることで、機能に影響が出ます。
新生突然変異(de novo変異)とは、両親には存在せず、お子さんで初めて生じた変異のこと。多くのFHM1は家族歴がありますが、孤発例の一部はこの新生突然変異が原因です。
代表的な再発性変異とその表現型
T666M変異(最頻)
世界中で最も頻繁に報告されるFHM1変異。重度の片麻痺性片頭痛と進行性小脳失調を高率に合併します。中国の家系報告では発端者が54歳時に発熱・昏睡・両眼の偏視を伴う急性脳症を発症、家系内7名全員がT666M陽性で、加齢に伴う失調の重篤化が確認されています。
R583Q変異
表現型に大きな不均一性があり、遅発性かつ永続的な進行性小脳失調と関連します。測定異常・企図振戦が目立ちます。さらに60歳以降に固縮・無動・安静時振戦といったパーキンソニズムを合併した症例も報告されており、R583Q特有の修飾因子の関与が示唆されています。
S218L変異
臨床的に最も危険な表現型を示します。日常的な片麻痺発作は経験しなくても、軽微な頭部外傷をきっかけに重篤な遅発性脳浮腫・神経脱落症状・致命的な昏睡を引き起こすリスクがあります。診断確定後は外傷予防の生活指導とスポーツ制限が不可欠です。
7. アレル疾患との鑑別診断——EA2・SCA6との連続スペクトラム
CACNA1A遺伝子の変異は、変異の種類(ミスセンス・ナンセンス・CAGリピート伸長など)によって異なる疾患カテゴリーを形成します。これらを「アレル疾患(Allelic disorders)」と呼び、FHM1のほかに反復発作性運動失調症2型(EA2)と脊髄小脳変性症6型(SCA6)が含まれます。臨床的にはベン図のように、片頭痛・発作性運動失調・進行性運動失調の各症状が連続したスペクトラムを形成しています。
| 疾患 | 変異の種類 | 発症年齢 | 主な臨床像 | 治療反応性 |
|---|---|---|---|---|
| FHM1 | ミスセンス(機能亢進) | 小児期〜若年期 | 運動麻痺を伴う重篤な片頭痛。半数で進行性慢性小脳失調を合併 | アセタゾラミドが一部に有効。トリプタンは禁忌 |
| EA2 | ナンセンス・スプライシング異常(機能喪失) | 小児期 | 数時間〜数日続く発作性運動失調とめまい。発作頻度が極めて高い。半数が片頭痛を伴う | アセタゾラミド・4-アミノピリジンが著効 |
| SCA6 | CAGリピート異常伸長(ポリグルタミン病) | 成人期後期(40〜50代以降) | 発作性症状を伴わない、緩徐進行性の純粋小脳失調 | 根本的治療なし。対症療法とリハビリ |
FHM1とEA2は臨床像のオーバーラップが多く、高度な神経内科医でも鑑別が容易ではありません。両者とも小児期発症で発作性の神経症状と慢性的な小脳失調を伴います。ただし意識障害を伴う重症発作はFHM1に特徴的で、EA2では稀。一方発作頻度はEA2が圧倒的に多く(年間中央値108回 vs FHM1の6.5回)、1時間未満の短いエピソードはEA2に特有です。確定診断には遺伝子検査が不可欠です。
8. 治療と長期管理
FHM1に対する根本的な治療法はまだ確立されていませんが、発作の予防と症状管理によって患者さんの生活の質(QOL)を大きく改善することは可能です。治療戦略は「片頭痛発作の管理」と「進行性小脳失調症の緩和」の二つに大別されます。
⚠️ 重要な禁忌:トリプタン製剤とエルゴタミン製剤
一般的な片頭痛の急性期治療に広く使われるトリプタン系薬剤(5-HT1B/1D受容体作動薬)とエルゴタミン製剤は、FHM1およびSHMにおいて絶対禁忌です。これらは強力な血管収縮作用を持つため、発作に伴う脳血流低下や虚血性脳卒中のリスクを不必要に増大させる可能性があります。遺伝子検査でFHM1と診断されることが、こうした致命的なリスクのある薬剤を避けるための最も確実な根拠になります。
片頭痛発作の治療
急性期治療
疼痛コントロールには非ステロイド性抗炎症薬(NSAIDs)・アセトアミノフェン・制吐薬が中心。長引く片麻痺や脳症を伴う重篤発作では、二次的なてんかん重積予防のため抗てんかん薬の静注と支持療法が必要です。
予防治療
皮質の過興奮を抑える抗てんかん薬が予防治療の主軸。トピラマート・バルプロ酸・クロナゼパムなどが選択肢となります。チャネル機能亢進という病態を直接ターゲットとする発想です。
進行性小脳失調症への薬物療法
💊 アセタゾラミド(ダイアモックス)
炭酸脱水酵素阻害薬で、アレル疾患であるEA2の発作予防薬として長らく第一選択。細胞内pHを下げることで神経細胞全体の過興奮を抑制すると考えられています。FHM1でも反復性の失調症状や前兆症状の重症度と頻度を軽減する効果が報告されています。
💊 4-アミノピリジン(4-AP)
カリウムチャネル阻害薬で、運動失調や眼振(特に下向き眼振)に対して非常に有効な対症療法薬。小脳プルキンエ細胞の電位依存性カリウムチャネルを阻害し、機能不全に陥ったプルキンエ細胞の静止発火の正確性を回復させます。推奨用量は5mgを1日2〜4回。動物モデルでは早期投与による神経保護作用も示唆されています。
その他の合併症への対応
失調性振戦にはプロプラノロール・プリミドン・トピラマート・ガバペンチンが、痙縮や筋ミオクローヌスにはクロナゼパムやバクロフェンが用いられます。小脳機能障害そのものに起因するうつ症状(Cognitive affective syndrome)も少なくないため、SSRI/SNRIによる薬物療法と心理サポートを並行して行います。理学療法・作業療法・言語嚥下療法を含む包括的リハビリテーションの早期導入が、長期予後を大きく左右します。
9. 遺伝カウンセリングと検査の選択肢
FHM1の確定診断後、また家族歴のある方が妊娠・出産を考える際には、丁寧な遺伝カウンセリングが重要な役割を果たします。常染色体顕性遺伝のため、患者さんから生まれるお子さんに50%の確率で遺伝する可能性があります。一方で、新生突然変異(de novo変異)による孤発例もあるため、家族歴の有無だけでは判断できません。
- ➤家系内既知変異が同定されている場合の出生前診断:絨毛検査・羊水検査によって胎児がCACNA1A変異を持つかを調べることが技術的には可能です。ただし表現型の幅が大きい疾患のため、検査前にご家族でじっくり話し合う時間が必要です。
- ➤包括的な単一遺伝子NIPTの選択肢:CACNA1Aを含む154遺伝子をカバーするインペリアルプランは、新生突然変異を含む単一遺伝子疾患の幅広いスクリーニングを目的とした検査メニューです。ご家族の状況や懸念に応じて選択できます。
- ➤診断後のサポート:診断確定により、トリプタン禁忌などの「使うべきでない薬」の明確化、S218L変異が見つかった場合の外傷予防の生活指導、進行性小脳失調症の早期介入計画——といった長期的なメリットが生まれます。遺伝カウンセリングのなかで丁寧に整理していきます。
- ➤診断・検査は強制ではない:検査を受けるかどうか、いつ受けるか、結果をどう生かすか——すべてご家族が自分のペースで決めることです。情報提供は医師の役割ですが、決定権はいつも患者さんとご家族にあります。
よくある質問(FAQ)
🏥 FHM1・希少遺伝性疾患のご相談
家族性片麻痺性片頭痛1型(FHM1)をはじめとする
CACNA1A関連疾患・希少遺伝性神経疾患のご相談は、
臨床遺伝専門医が在籍するミネルバクリニックへ。
関連記事
参考文献
- [1] Jen JC. Familial Hemiplegic Migraine. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. [NCBI Bookshelf NBK1388]
- [2] OMIM #141500. Migraine, Familial Hemiplegic, 1; FHM1. Johns Hopkins University. [OMIM]
- [3] Orphanet. Familial or sporadic hemiplegic migraine. ORPHA:569. [Orphanet]
- [4] ICHD-3. 1.2.3.1.1 Familial hemiplegic migraine type 1 (FHM1). International Headache Society. [ICHD-3]
- [5] Ducros A. Familial and sporadic hemiplegic migraine. Rev Neurol (Paris). 2014. [IHS]
- [6] Pelzer N, et al. Familial Hemiplegic Migraine With Progressive Cerebellar Ataxia Caused by a p.Thr666Met CACNA1A Gene Mutation in a Chinese Family. Front Neurol. 2019. [PMC6882281]
- [7] R583Q CACNA1A variant in SHM1 and ataxia: case report and literature update. J Headache Pain. 2012. [PMC3381060]
- [8] Pelzer N, et al. Diagnostic and therapeutic aspects of hemiplegic migraine. J Neurol Neurosurg Psychiatry. 2020. [PMC7361005]
- [9] Strupp M, et al. Aminopyridines for the treatment of neurologic disorders. Neurol Clin Pract. 2017. [PMC5310209]
- [10] Indelicato E, Boesch S. From Genotype to Phenotype: Expanding the Clinical Spectrum of CACNA1A Variants in the Era of Next Generation Sequencing. Front Neurol. 2021. [Frontiers]






