目次

第8番染色体異常のすべて:トリソミー・部分モノソミーの原因から症状、最新診断まで
📍 クイックナビゲーション
「第8番染色体の異常」という見慣れない診断名や検査結果を前に、深い戸惑いと不安を抱えてこの記事にたどり着かれたことと思います。ネット上には断片的な情報が溢れ、調べれば調べるほど絶望的な気持ちになってしまうご家族を、私はこれまでのべ10万人以上の方々の意思決定に伴走する中で数え切れないほど見てきました。
第8番染色体の異常は、染色体全体が関わる「トリソミー」から、微小な部分が欠けたり重複したりする「構造異常」まで、その症状や予後は極めて多様です。決して「ひとつの絶望的なストーリー」に当てはまるわけではありません。この記事では、臨床遺伝専門医の立場から、医学的なメカニズムから具体的な症状、そして「これからどう向き合っていくべきか」という未来への道筋まで、正確な事実をわかりやすく、そして誠実にお伝えします。
Q. 第8番染色体異常とはどのような状態ですか?
A. 染色体の数や構造に変化が生じることで、様々な合併症を引き起こす疾患群です。
第8番染色体は中枢神経や心臓、骨格の形成に重要な役割を持ちます。染色体全体が3本になる「トリソミー8」はモザイク型(正常細胞と混在)のみが生存可能であり、一部が欠ける「部分モノソミー(微小欠失)」は影響を受ける遺伝子の場所によって全く異なる隣接遺伝子症候群を引き起こします。
- ➤トリソミーのメカニズム:完全型が致死となる理由と、生存を可能にする「モザイク現象」
- ➤短腕(8p)の異常:逆位重複欠失8p症候群や、心疾患を伴う8p23.1欠失の症状
- ➤長腕(8q)の異常:複数の遺伝子が関与するTRPS II(毛髪鼻指節症候群II型)などの特徴
- ➤検査と診断:偽陽性が多いワイドゲノム法を避け、ターゲット法による正確なNIPTを選ぶ重要性
1. 第8番染色体の役割と「数的異常(トリソミー)」のメカニズム
人間の細胞には通常23対(合計46本)の染色体が存在します。そのうちの「第8番染色体」は、約1億4,600万塩基対という長大なDNAで構成され、約700個の遺伝子が乗っています。これらの遺伝子は、中枢神経系の発生、骨格の形成、心臓の器官形成など、胎児が成長するための極めて重要なプロセスを制御しています。
染色体異常には大きく分けて、染色体そのものの数が増減する「数的異常」と、一部が欠けたり重複したりする「構造異常」があります。まずは、数的異常である「トリソミー8」について、その医学的メカニズムを解説します。
受精の段階で誤って3本になってしまった染色体を、細胞分裂の過程で偶然に1本喪失し、正常な2本(二倍体)に戻ろうとする細胞の自己修復機能のことです。この現象により、正常な細胞と異常な細胞が混ざり合う「モザイク状態」が生み出されます。
完全トリソミーの致死性とモザイクによる生存(ワルカニー症候群2)
もし、すべての細胞で第8番染色体が3本存在する「完全トリソミー」が発生した場合、遺伝子の過剰発現が極めて重篤な影響を及ぼし、事実上、生命維持と両立しません。完全な第8番染色体トリソミーは、妊娠のごく初期段階で致死的(自然流産)となります。
しかし、現実には第8番染色体が3本ある状態で生まれてくるお子様がいらっしゃいます。これは例外なく、上述の「トリソミー・レスキュー」や、受精後の細胞分裂エラーによって、正常な細胞(2本)とトリソミー細胞(3本)が混ざり合った「モザイク型(モザイク・トリソミー8)」となっているためです。正常な二倍体細胞系統を獲得し、重要臓器の発生を補償することで胎児の生存が可能となります。この状態は発見者の名にちなみ「ワルカニー症候群2」とも呼ばれます。
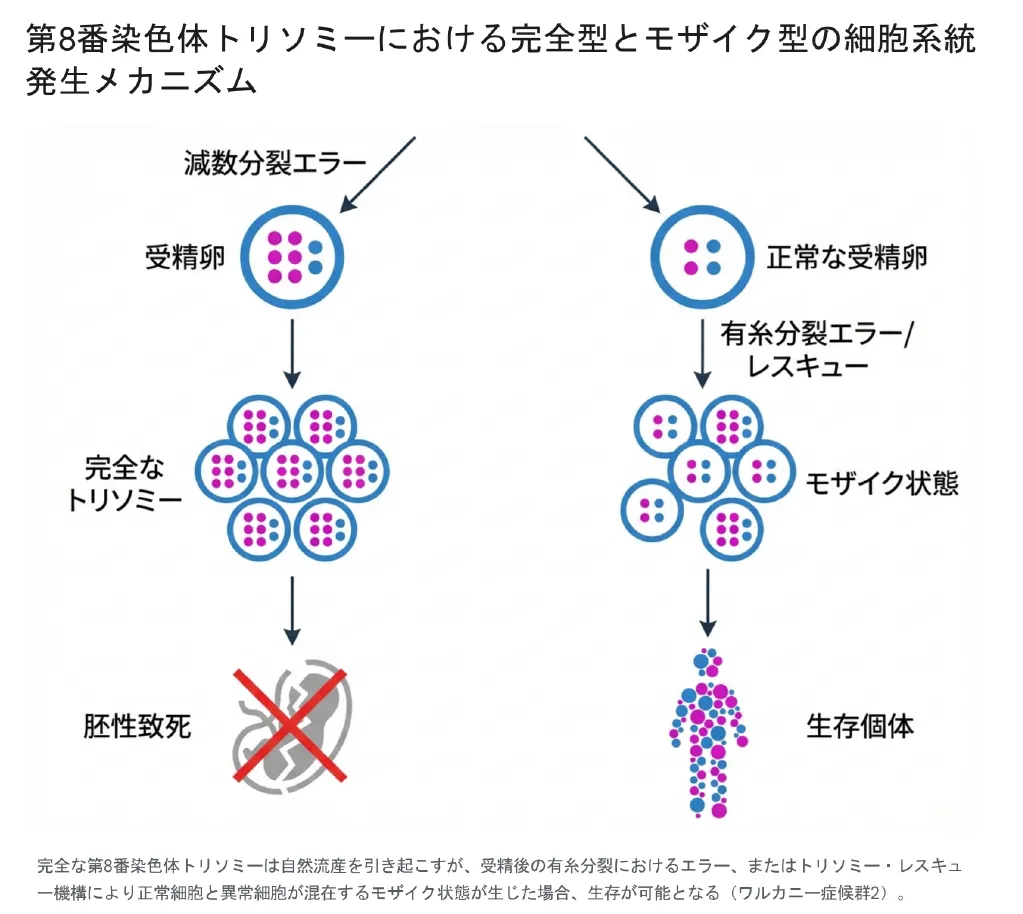
完全型とモザイク型の発生メカニズムの違い
実際の診療現場におけるケーススタディ:見逃されやすい「組織特異的モザイク」
臨床現場で私たちが直面する大きな課題の一つが、「組織特異的なモザイク現象(Tissue-specific mosaicism)」による診断の難しさです。
ある患者様は、発達の遅れや特徴的な顔貌、関節の拘縮など、ワルカニー症候群2を疑う強い所見がありました。しかし、一般的な末梢血(血液)を用いた染色体検査では「正常(異常なし)」という結果が出たのです。血液の細胞は入れ替わり(ターンオーバー)が激しいため、増殖に不利な異常細胞が淘汰されてしまい、血液中からはトリソミー細胞が消えてしまうという現象(偽陰性)がしばしば起こります。
そこで、患者様の皮膚を注意深く観察したところ、「ブラシュコ線」と呼ばれる渦巻状の色素沈着が見られました。皮膚生検を実施し、皮膚の線維芽細胞を培養して再検査を行った結果、高い割合でトリソミー8の細胞が検出され、確定診断に至りました。
💡 専門医からのアドバイス:血液検査の限界を知る
「血液検査で異常がなかったから絶対に大丈夫」と思い込み、根本的な原因がわからないままリハビリや療育に苦労されているご家族がいます。症状があるのに原因が不明な場合、組織特異的モザイクを熟知した臨床遺伝専門医の評価を受けることが、正確な診断と適切な支援に繋がる第一歩です。
2. 第8番染色体短腕(8p)の構造異常:微小欠失と重複がもたらす影響
染色体の異常は、全体の数が増えるだけでなく、一部分が欠ける(欠失/部分モノソミー)ことや、余分に付着する(重複/部分トリソミー)ことでも起こります。特に第8番染色体の「短腕(8p)」と呼ばれる領域は、ゲノム構造が非常に脆いという特徴があります。
この領域には、似たようなDNA配列(嗅覚受容体遺伝子のクラスター)が並んでおり、親の生殖細胞が作られる際(減数分裂時)に、誤った位置でDNAが交差してしまう「非対立遺伝子間相同組換え(NAHR)」が頻発します。これが、複雑な構造異常を引き起こす医学的なメカニズムです。
通常、遺伝子は両親から1つずつ(計2コピー)受け継ぎます。微小欠失などによって1つのコピーが失われ、残った1つの遺伝子だけでは必要なタンパク質の量を十分に作り出せず、正常な機能が維持できなくなって病気を発症する状態を指します。
逆位重複欠失8p症候群と8p23.1欠失症候群
8p領域の異常による代表的な疾患に、**逆位重複欠失8p症候群(Inv Dup Del 8p)**があります。これは、染色体の末端が欠失し、中間部分が逆向きに重複するという極めて複雑な「新生突然変異(de novo:親からの遺伝ではなく、受精卵ができる過程で偶然生じる変異)」です。脳梁の欠損や、乳幼児期の筋緊張低下から進行性の過緊張への移行、自閉症スペクトラム障害(ASD)やADHDなどの強い行動的特徴が見られます。
また、**8p23.1欠失症候群**(詳しくは8p23.1微細欠失症候群をご覧ください)は、心臓の正常な形成に不可欠な「GATA4遺伝子」が失われる(ハプロ不全に陥る)ことで発症します。患者の75%〜94%に、房室中隔欠損症などの重篤な先天性心疾患が合併し、生命予後に直結するため、出生直後からの高度な循環器サポートが必須となります。一方で、同じ領域のコピー数が増える8p23.1微細重複症候群も存在し、欠失とは異なる臨床的特徴を持ちます。
実際の診療現場におけるケーススタディ:行動の背景にある遺伝的要因
Inv Dup Del 8pの診断を受けたある男の子のケースです。ご両親は「かんしゃくがひどく、コミュニケーションが取れない。私たちの育て方が悪いのか」と深くご自身を責めていらっしゃいました。
しかし、この行動特性の根底には、欠失・重複した8p領域に含まれる社会性や情動に関連する特定の遺伝子(MCPH1、DPYSL2など)の増減が、脳の神経回路網の発達に複合的な影響を及ぼしているという明確な医学的根拠があります。この事実をお伝えしたことで、ご両親は「育て方のせいではなかった」と涙を流され、そこから「この子の染色体の特性に合わせた環境作り(代替的コミュニケーション手段の導入や応用行動分析)」へと前向きに舵を切ることができました。

3. 第8番染色体長腕(8q)の構造異常:TRPS IIと組換え第8番染色体症候群
第8番染色体の異常は長腕(8q)でも発生します。ここでのキーワードは「隣接遺伝子欠失症候群(Contiguous Gene Deletion Syndrome)」です。これは、染色体上で物理的に隣り合っている複数の異なる遺伝子が、一つの微小欠失イベントによって「まとめて」失われることで、複数の独立した症状が組み合わさって現れる疾患概念です。
8q23.3-q24.11領域の欠失によって生じます。TRPS1遺伝子の喪失が「特異な顔貌や細い毛髪」を、EXT1遺伝子の喪失が「多発性外骨腫(良性の骨腫瘍)」を、RAD21遺伝子のハプロ不全が「知的障害」を引き起こすというように、欠失した遺伝子と症状が明確に一対一で対応しているのが特徴です。
この疾患についてより深く知りたい方は、8q24.11–q24.13欠失症候群(Langer-Giedion症候群/TRPS2)や、NIPTでの検査対象となる8q23q24欠失症候群の解説ページも併せてご参照ください。
また、8q領域には他にも、8q21.11欠失症候群や、特徴的な顔貌を呈する8q22.1微細欠失症候群(ナブルスマスク様顔面症候群)など、欠失する領域(メガベース単位の位置)によって全く異なる特異的な隣接遺伝子症候群が報告されています。
組換え第8番染色体症候群(サン・ルイス・バレー症候群)
もう一つ、非常に特殊な成り立ちを持つのが「組換え第8番染色体症候群」です。これは、親が第8番染色体に「挟腕部逆位(遺伝子の順番が180度反転しているが、遺伝物質の増減がないため親自身は健康な状態)」を持っている場合に起こります。
親の生殖細胞が形成される際の減数分裂で、不均等な交差が起こり、結果として子には「8p末端の欠失」と「8q末端の重複」が同時に伝達されてしまいます。ファロー四徴症などの致死的な先天性心疾患が頻発するため、早期の外科的介入が不可欠です。
実際の診療現場におけるケーススタディ:症状の数に圧倒されないための「集学的アプローチ」
TRPS IIの診断を受けた患者様のご家族は、骨格の変形、顔貌の特徴、知的発達の遅れなど、次々と提示される症状のリストに圧倒され、強い恐怖を抱いていました。特にEXT1遺伝子の欠失による「多発性外骨腫」は、骨の表面から外側に向かって多数の良性腫瘍が成長し、激しい痛みや運動障害を引き起こします。
このような場合、単一の診療科だけで問題を解決することは不可能です。私はご家族に対し、「これらすべてを一度に治す魔法はありませんが、一つひとつの症状に対して専門家がチームを組みます」とお伝えしました。集学的医療モデルを構築(整形外科による外骨腫の定期的な切除とペインコントロール、小児内分泌科でのホルモン管理、そして特別支援教育の導入など)することで、患者様は痛みを抑えながら学校生活を楽しむことができるようになりました。
4. 出生前診断と出生後診断:NIPT(第3世代)と最新ゲノム解析の役割
ここで極めて重要な大原則をお伝えします。医療における診断には、「出生前診断(生まれる前の検査)」と「出生後診断(生まれた後の確定検査)」の明確な分離があります。「診断」という言葉がすべて出生前に行われると誤解されている方も多いですが、出生前に行えるのは原則として羊水検査や絨毛検査などの侵襲的検査によって得られる確定診断のみであり、採血によるNIPTはあくまで「スクリーニング(非確定検査)」です。
当院が提供する「ダイヤモンドプラン」の真価
ミネルバクリニックでは、非認証施設でありながら、臨床遺伝専門医が遺伝カウンセリングから判定、陽性後のケアまで一貫して行う、国内でも極めて稀有(rare)な医療体制を敷いています。
特に、最もカバー範囲が広く多くの方に選ばれている「ダイヤモンドプラン」は、SNP法とターゲット法を融合させた最先端の「COATE法」を採用しています。これにより、常染色体トリソミー(13, 15, 16, 18, 21, 22)、性染色体異数性(4種)、そして12領域の微細欠失(1p36, 4p16など)に対して、陽性的中率「>99.9%」という極めて高い精度を誇ります。微細欠失の積算リスクは一般的な妊婦さんでも1/1000存在するため、この精度の高さはご家族にとって最大の安心材料となります。
さらにダイヤモンドプランでは、父親由来の精子の新生突然変異に起因する「単一遺伝子(56遺伝子)」の検査も網羅しています。この積算リスクは1/600(当院のデータでは1/60人が陽性)であり、行動・知的発達に重い影響を及ぼし、重度の合併症を伴う「症候性(重症)自閉症」の原因となる変異も多数含まれています。
(父親由来の遺伝子変異が子へ伝わるイメージ)
出生後の確定診断:マイクロアレイ染色体検査(CMA)の役割
出生後に症状が見られた場合、あるいは羊水検査において微細な構造異常を疑う場合(※学会指針では原則として超音波での構造異常がある場合などが対象)、従来の光学顕微鏡を用いた「Gバンド法」では、TRPS IIや8p23.1欠失症候群のような微小な欠失を見つけることは困難です。
現在では、数キロベース単位の解像度でゲノム全体のコピー数変異(CNVs)を網羅的に検出できる「マイクロアレイ染色体検査(CMA)」が、出生後診断のゴールドスタンダードとなっています。これにより、正確なゲノムの切断点と同定が可能となり、予後の予測が飛躍的に進歩しました。
5. 長期的な臨床管理とサポート:診断はゴールではなくスタート
第8番染色体の異常(数的・構造的)に対する、遺伝子レベルでの根本的な治療法は現時点では確立されていません。しかし、だからといって「何もできない」わけでは決してありません。
特に、モザイク・トリソミー8(ワルカニー症候群2)の患者様における重要な管理ポイントの一つが「発がんリスクのモニタリング」です。先天的なトリソミー細胞がプレレウケミック状態(白血病の前段階)として機能し、急性骨髄性白血病(AML)などの特定の血液悪性腫瘍を発症するリスクが一般集団より有意に高いことが分かっています。そのため、血液内科医による定期的な全血球計算(CBC)検査を中心とした慎重な長期的モニタリング体制を維持することが、長期的な予後の改善において決定的な意味を持ちます。
ネットの断片的な情報に疲れ果ててしまったら、一人で抱え込まず、一度専門医の話を聞きに来ませんか。当院では互助会(8,000円)により、羊水検査費用が全額補助される安心の仕組みも整えており、2025年6月からは院内での確定検査(羊水・絨毛検査)も可能となります。不安な時間を最小限に抑え、確かな事実に基づいた「これからの選択」を共に考えていきましょう。
🔍 関連記事
よくある質問(FAQ)
関連記事
参考文献
- [1] Chromosome 8 – MedlinePlus Genetics [MedlinePlus]
- [2] Recombinant 8 syndrome – MedlinePlus Genetics [MedlinePlus]
- [3] Trisomy 8 Mosaicism – RareChromo.org [RareChromo]
- [4] Trichorhinophalangeal syndrome type II – Genetics – MedlinePlus [MedlinePlus]
- [5] 8p inverted duplication/deletion syndrome – Orphanet [Orphanet]






