目次
11q23-q24.3欠失症候群(Jacobsen症候群)
この記事の著者 仲田洋美(総合内科専門医、がん薬物療法専門医、臨床遺伝専門医)
11q23.3-q24.2領域の新生突然変異欠失
抄録
背景
Jacobsen症候群(JBS)は11q末端欠失が関与する隣接遺伝子欠失症候群である。遠位11qにおける欠失はまれであり、JBSの臨床的表現型へのそれらの寄与は不明である。
症例提示
11qの欠失領域と表現型の間の相関を解析するために、11q23.3‐q24.2領域にそれぞれ重複しないde novo欠失を有する2人の個体の染色体マイクロアレイ(CMA)データと臨床的特徴を提示した。どちらの欠失も患者の状態に病原性を示す可能性が高い。11q23.3q24.1の欠失は、低身長、相対的な小頭症、発育不全、低張および睡眠障害と関連している。11q24.2での欠失はHEPACAMを伴い、著者らの患者の臨床症状(相対的大頭症、異常MRI、軽度発達遅延および発作)は、皮質下囊胞2Bを伴う中脳白質脳症と矛盾しない。
結論
本発明者らは、11q23.3−qterにおける1つより多くの重要領域がJBSの可変臨床症状の原因であり、したがって、Jacobsen症候群(JBS)は、複数の遺伝子座がJBSの臨床特性に寄与した真の連続遺伝子欠失症候群であるという概念を支持することを見出した。11q23.3-q24.2での小さな欠失およびそれらに関連する独特の特徴もまた、新たなゲノム障害の出現を示唆する。
背景
Jacobsen症候群(OMIM #147791)としても知られる11q末端欠失は、11qterの欠失を含む隣接遺伝子欠失症候群である。Jacobsenが1973年に初めて報告して以来、11q末端欠失患者数百例が記載されていた[1]。JBS患者において観察される欠失は、7~20Mbの範囲の大きさであり、そしてほとんど常に11q末端欠失と関連し、そして最も遠い切断点は、11q23.3に位置する。JBSの典型的な特徴には、発達遅滞(DD)/知的障害(ID)、異形顔貌、血小板障害および多発性先天性欠損がある[1~6]。11q23から11qterまでの欠失はまれであり、その臨床的意義は現在のところ不明である。以前に報告された11q23-qter欠失の一部は、核型分析により特徴付けられた[7~9]。CMA解析は、遺伝子型‐表現型相関をより明確にすることを可能にした。CMA [10]により特徴付けられた11q24.1q24.3性欠失の最初の報告以降、CMAにより検出された11q23-qter性欠を有する8例が文献に記録されている(追加ファイル1:表S1参照)。11q23-qter欠失の切断点は様々であり、欠失の大きさは2.89~12.8Mbである。罹患者の臨床像も非常に不均一であるため、遺伝子型-表現型相関分析が複雑になっている[4,5,10-14]。11q23-qter領域に性欠失を有する患者は、低緊張[9]、大頭症[11,14]、小頭症[5]、三角頭症[4,7,9]、異形顔貌の一部[4,5,7-14]、四肢奇形[7,11,13]、先天性心欠損(CHD) [4,5,7,9,10,12]などのJBSの特徴の一部を共有することができたが、JBSの全体集合を示さないことが多かった[13,14]。11q23-qterの欠失を認める全患者において最も一致した特徴は、DD/IDである。最小の重複領域は、いくつかの重要領域または遠位11q領域の候補遺伝子を定義するのに役立った[4~11]。
ここでは、遺伝子型‐表現型相関をさらに定義するために、異なる程度のDD/IDと幾つかの異形性特徴を有する11q23.3‐q24.2領域で重複しない欠失を有する2人の個体を報告した。
症例提示
患者1
患者1(P1)は在胎41週齢の29歳女性から生まれた5歳女児であった。妊娠、分娩は順調であった。出生体重は2.8kg(~15th百分位数)、出生体長は48.2cm(~15th百分位数)であった。彼女は外見上健康な双生児の兄弟と姉がいた。生後5~6か月頃に、転がり過ぎない、這い回りが遅れる、座る、立つなどの全般的な遅れがあることに気づいた。1歳時に5カ月の発達レベルであった。発育不全と筋緊張低下の既往があった。異形性の特徴と全般的な発達遅滞によりGenetics Clinicに来院した。5歳時、身長101cm(-2.11 SD)、体重14kg(-2.58 SD)、頭囲48.3cm (7パーセンタイル)であった。足趾の重複、軽度の遠視症(内眼角距離27mm、+1SD)、前額部の突出、顔面の平坦プロフィール、広鼻、上唇が薄くてなめらかな好気性、上眼角ヒダを伴う上傾性眼瞼裂を含む異形性の特徴を有していた。脳MRIは正常であった。他に欠損は認めなかった。
睡眠障害の既往があった。とりわけ、彼女の両親は就寝前に発声すると報告しており、しばしば身体の揺れや頭部の打撲と関連している。彼女は目を覚まし、真夜中に頭を叩いた。彼女はベッドの上にインド風に座り、マットレスに頭をぶつけるまで身を乗り出した。これはかなり速くリズミカルなパターンで起こった。そのような動きの間、彼女はしばしば母親を嘆き、何度も母親は非常に大きな叫び声にエスカレートした。また、日中、背中を硬い表面に当てて動かし、その後、腰と臀部を床に動かすような揺動行動をしていた。夜間の頭部白せんと診断し、このような行動は改善したが完全には消失しなかった。
患者2
患者2(P2)は10か月齢時に発熱、軽度発達遅延および発作のため来院した男児であった。健常な親から満期第2子で、健常な4歳の娘がいた。妊娠中、出産中に合併症なく帝王切開で出産した。出生時体重は3.25kg (25~50パーセンタイル)、出生時体長は50cm (25~50パーセンタイル)であった。来院前に重大な既往医療かった。発作、自閉症、精神遅滞または他の神経学的障害の家族歴はない。生後3~4カ月頃に頭を上げ、生後6~7カ月頃に転がり、生後10カ月頃に初発語を顕著にした。彼の開発指数(DQ)と精神指数(MI)は、デンバー開発スクリーニングテストスコア(DQ = 51とMI = 63)により軽度の開発遅延があることを示唆した。生後10か月時の体重は9.4kg (30パーセンタイル)、身長は72.5cm (25パーセンタイル)であった。相対的大頭症(周囲径47.5cm (+1.3SD))、軽度の高眼圧症、低鼻梁、薄い上唇および斜視を呈した。磁気共鳴画像(MRI)は、前頭側頭葉の拡大、完全な両側脳室および深頭頂溝を明らかにした。脳波、筋電図は正常であった。急性咽頭炎、二次性発作、精神運動遅滞および軽度の発達遅延ならびに脳低形成と診断された。
方法
染色体マイクロアレイ(CMA)解析
標準プロトコールを用いて末梢血リンパ球からDNAサンプルを抽出し、患者1にはAgilent 244 Kアレイを、患者2には照明器HumanSNP cyto-12アレイを用いてマイクロアレイを行った。
結果
11q23.3‐q24.1(chr11:120410050‐122085906) (hg19)の領域での1.6Mb欠失がP1のゲノムで検出された。11q24.2(chr11:124635144-125390604) (hg19)における0.76Mbの欠失がP2のゲノムに検出された(図1)。親のCMAは、両方の欠失がde novoであることを示した。
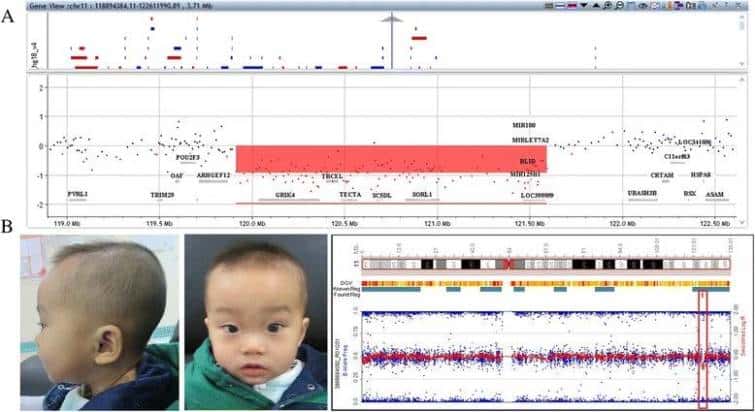
P1における11q23.3-q24.1の欠失(赤色で影を付けた)は、Agilent 4X180Kアレイによって検出された。b P2の顔貌。前額部が大きい、軽度の高眼圧症、鼻梁が低い、上唇が薄い、斜視。イルミネーションアレイは、11q24.2での小さな欠失(赤いレクタングル)を検出した。
考察
11qの欠失はまれであり、遺伝子型-表現型研究は、報告された症例数が少なく、患者の臨床的表現型に関する十分な詳細が不足していたために妨げられていた。これまでのところ、11q23.3-q25領域に11の欠失症例しか報告されていない(追加ファイル1:表S1)。そのうち8例について染色体マイクロアレイデータが利用可能であった。Guerinらは、11q24.2-q24.3に2.899Mbの欠失が認められ、三角頭症、毛様体機能亢進症、深部眼を呈した患者[4]、11q25に4.11Mbの欠失が認められ、重度のDD、小頭症、顔面奇形を呈した患者[5]、11q24.2q24.3に4.74Mbの欠失が認められ、大頭症、ID、および異常なMRIを呈した患者[11]、SO Jらは、11q24.2-q24.3に3.162Mbの欠失が認められ、心室周囲結節性異所性および横断肢縮小欠失が認められた女性を報告した[13]、Yamamotoらを報告した 11q23.3q24.2に5.3Mbの欠損を認め、出生前の大頭症および軽度の発達遅延を呈した20か月齢の男児[14]。ここで報告した2名の患者がこのリストに追加された。P1は低身長、DD/ID、相対的小頭症、睡眠障害、幾つかの異形性の特徴を呈し、P2は軽度のDD/ID、相対的大頭症、軽度の高眼球症を示した。以前に報告された11q23-qterの欠失を有する患者8人および現在の症例2人の臨床的特徴の要約を表1に示す。これらの欠失患者における表現型の有病率は少数では定量化できないが、まとめは、ヤコブセン症候群は欠失領域内に関与する複数の遺伝子(領域)が症候性表現型に役割を果たす隣接遺伝子欠失症候群であるという概念を総合的に支持している。
表1
11q23-qter欠失8例の臨床像のまとめと自験例
| 臨床所見 | 以前の症例 | P1 | P2 |
| 番号 | 8 | 1 | 1 |
| 地域 | 11q23-q25 | 11q23.3q24. | 11q24.2 |
| 欠失サイズ(Mb) | 2.89–12.8 | 1.6 | 0.76 |
| 性別 | 3 m/5f | f | m |
| 年齢 | 新生児から成人までの範囲 | 4 years | 10 months |
| 出生時体重 | 低正常 | ~15% | 25~50% |
| 低緊張症 | – | + | – |
| 大頭症 | 2 | – | 相対的大頭症 |
| 小頭症 | 2 | 相対的小頭症 | – |
| 三角頭症 | 1 | – | – |
| 顕著な額 | 1 | + | – |
| 高テロリズム | 2 | 軽度 | 軽度 |
| 眼瞼裂異常 | 4 | + | – |
| 耳の奇形 | 4 | – | – |
| 鼻の奇形 | 3 | + | + |
| 口腔奇形 | 3 | + | + |
| 四肢の奇形 | 2 | + | – |
| 心血管系の異常 | 4 | – | – |
| 血液学的異常 | 3 | – | – |
| 発達遅滞/知的障害 | 6 | + | + |
| 社会的交流の困難 | 2 | – | NA |
臨床所見:+、存在;-、欠如、f雌、m雄、NA非該当DD/IDはJBSの一貫した特徴である。また、11qの欠失を有する患者によくみられる特徴である(Table 1)。Taoyun Jiらは、4.1Mb欠失で定義されるテロメア近傍のDD/IDの臨界領域を提唱した(図2の症例3)[5]。我々のP2で検出された欠失はサイズが最も小さく、症例1[13]および4[11]で検出された欠失と重複している。3例ともDD/IDの臨床症状を示し、この観察に基づき、11q24.2(chr11:124635144-125390604)に新規DD/ID遺伝子座を提案した。同様に、本発明者らのP1において検出された欠失は、11q23.3−q24.1(chr11: 120410050−122085906)における別の新規なDD/ID遺伝子座を規定し得る。したがって、11q上には、各領域のハプロ不全がDD/IDと関連している可能性が高い複数の遺伝子座が存在する。
 図 2
図 2報告された11q23-qter領域症例(UCSC Genome Browser, hg19)における欠失を指す模式図
患者1がリズミカル運動障害(RMD)としても知られる、夜間頭部黄疸の特徴を示したことは興味深い。RMDのほとんどは4歳までに自然消失する[15]。患者の状態は改善したが、5歳で完全には消失しなかった。RMDの根本的な原因は現時点では未知であるが、P1における欠失はRMDの分子メカニズムを調査する手がかりになるかもしれない。10のOMIM遺伝子(GRIK4、LRC35、TECTA、SC5DL、SOR1、MIR100HG、MIR125B1、BLID、MIRLET7A2、MIR100)を含むRef13 Seq遺伝子は欠失に関与している。しかし、どの遺伝子または遺伝子が睡眠障害の原因である可能性が高いかは不明である。GRIK4(OMIM #600282)はグルタミン酸依存性イオンチャネルファミリーに属するタンパクをコードする。グルタミン酸は、リガンド依存性イオンチャネルおよびGタンパク共役膜受容体の活性化を介して、中枢神経系における主要な興奮性神経伝達物質として機能する。Takenouchi T et al.およびPickard BS et al.は、GRIK4のハプロ不全がDD、精神遅滞、統合失調症および双極性障害に関連していることを示唆した[16,17]。著者らの患者に存在する発達遅延は、GRIK4欠失によって説明される可能性がある。
P2における欠失間隔は6つのOMIM遺伝子(ROBO3、ROBO4、HEPACAM、HEPN1、PKNOX2およびFEZ1)を包含した。HEPACAMにおける配列変異体(OMIM #611642)は、皮質下囊胞2A(常染色体劣性型MLC2A、OMIM #613925)および2B(常染色体優性型MLC2B、OMIM #613926)を伴う巨脳性白質脳症を引き起こすことが示されており、いずれも大頭症、MRI異常および様々な程度の知的障害を特徴とする[18~20]。常染色体型の臨床像はより軽度で、一部の特徴は年齢とともに改善する。最近、HEPACAMのハプロ不全は、11q23.3q24.2欠失11q24およびMLCの臨床的特徴でヘテロ接合性欠失を有する2人の患者の原因と考えられた[14]。P2は相対的大頭症、MRI異常軽度発達遅延および発作を呈したが、MLC2Bとは一致しなかった。P2で検出されたはるかに小さな欠失は、山本の報告の患者2の欠失と重複している。この2症例間の最小重複領域は、軸索の伸長に役割を果たし、Tysonら[11]によりMRI異常の候補遺伝子として提唱されているFEZ1(OMIM #604825)遺伝子の関与を除外することができる。
結論
要約すると、著者らはJBS領域における2つの稀なde novo欠失について述べた。ID/DDはJBSと共通の特徴であり、ヤコブセン症候群は真の隣接遺伝子欠失症候群であり、ID/DDの重要領域は11q末端の異なる領域に存在するという概念を支持する。著者らの研究はさらに、DD/IDと関連する最小臨界領域を定義した。また、各欠失は、その独特の特徴を示し、明確な新規ゲノム不均衡障害を示唆した。
- Mattina T, Perrotta CS, Grossfeld P. Jacobsen syndrome. Orphanet J Rare Dis. 2009;4:9. doi: 10.1186/1750-1172-4-9. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Grossfeld PD, Mattina T, Lai Z, et al. The 11q terminal deletion disorder: a prospective study of 110 cases. Am J Med Genet A. 2004;129A(1):51–61. doi: 10.1002/ajmg.a.30090. [PubMed] [CrossRef] [Google Scholar]
- Penny LA, Dell Aquila M, Jones MC, et al. Clinical and molecular characterization of patients with distal 11q deletions. Am J Hum Genet. 1995;56(3):676–83. [PMC free article] [PubMed] [Google Scholar]
- Guerin A, Stavropoulos DJ, Diab Y, et al. Interstitial deletion of 11q-implicating the KIRREL3 gene in the neurocognitive delay associated with Jacobsen syndrome. Am J Med Genet A. 2012;158A(10):2551–6. doi: 10.1002/ajmg.a.35621. [PubMed] [CrossRef] [Google Scholar]
- Ji T, Wu Y, Wang H, Wang J, Jiang Y. Diagnosis and fine mapping of a deletion in distal 11q in two Chinese patients with developmental delay. J Hum Genet. 2010;55(8):486–9. doi: 10.1038/jhg.2010.51. [PubMed] [CrossRef] [Google Scholar]
- Fryns JP, Kleczkowska A, Buttiens M, Marien P, van den Berghe H. Distal 11q monosomy. The typical 11q monosomy syndrome is due to deletion of subband 11q24.1. Clin Genet. 1986;30:255–60. doi: 10.1111/j.1399-0004.1986.tb00605.x. [PubMed] [CrossRef] [Google Scholar]
- Sirota L, Shabtai F, Landman I, et al. New anomalies found in the 11 q- syndrome. Clin Genet. 1984;26:569–73. doi: 10.1111/j.1399-0004.1984.tb01105.x. [PubMed] [CrossRef] [Google Scholar]
- Ono J, Hasegawa T, Sugama S, et al. Partial deletion of the long arm of chromosome 11: ten Japanese children. Clin Genet. 1996;50:474–8. doi: 10.1111/j.1399-0004.1996.tb02715.x. [PubMed] [CrossRef] [Google Scholar]
- Pivnick EK, Velagaleti GV, Wilroy RS, et al. Jacobsen syndrome: report of a patient with severe eye anomalies, growth hormone deficiency, and hypothyroidism associated with deletion 11 (q23q25) and review of 52 cases. J Med Genet. 1996;33(9):772–8. doi: 10.1136/jmg.33.9.772. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Wenger SL, Grossfeld PD, Siu BL, et al. Molecular characterization of an 11q interstitial deletion in a patient with the clinical features of Jacobsen syndrome. Am J Med Genet A. 2006;140(7):704–8. doi: 10.1002/ajmg.a.31146. [PubMed] [CrossRef] [Google Scholar]
- Tyson C, Qiao Y, Harvard C, Liu X, et al. Submicroscopic deletions of 11q24-25 in individuals without Jacobsen syndrome: re-examination of the critical region by high-resolution array-CGH. Mol Cytogenet. 2008;1:23. doi: 10.1186/1755-8166-1-23. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Van Zutven LJ, van Bever Y, Van Nieuwland CC, et al. Interstitial 11q deletion derived from a maternal ins(4;11)(p14;q24.2q25): a patient report and review. Am J Med Genet A. 2009;149A(7):1468–75. doi: 10.1002/ajmg.a.32714. [PubMed] [CrossRef] [Google Scholar]
- So J, Stockley T, Stavropoulos DJ. Periventricular nodular heterotopia and transverse limb reduction defect in a woman with interstitial 11q24 deletion in the Jacobsen syndrome region. Am J Med Genet A. 2014;164A(2):511–5. doi: 10.1002/ajmg.a.36292. [PubMed] [CrossRef] [Google Scholar]
- Yamamoto T, Shimada S, Shimojima K, et al. Leukoencephalopathy associated with 11q24 deletion involving the gene encoding hepatic and glial cell adhesion molecule in two patients. Eur J Med Genet. 2015 Sep;58(9):492–6. [PubMed]
- Khan A, Auger RR, Kushida CA, et al. Rhythmic movement disorder. Sleep Med. 2008 Mar;9(3):329–30. [PubMed]
- Takenouchi T, Hashida N, Torii C, et al. 1p34.3 deletion involving GRIK3: Further clinical implication of GRIK family glutamate receptors in thepathogenesis of developmental delay. Am J Med Genet A. 2014;164A(2):456–60. doi: 10.1002/ajmg.a.36240. [PubMed] [CrossRef] [Google Scholar]
- Pickard BS, Malloy MP, Christoforou A, et al. Cytogenetic and genetic evidence supports a role for the kainate-type glutamate receptor gene, GRIK4, in schizophrenia and bipolar disorder. Mol Psychiatry. 2006;11(9):847–57. doi: 10.1038/sj.mp.4001867. [PubMed] [CrossRef] [Google Scholar]
- Lopez-Hernandez T, Ridder MC, Montolio M, Capdevila-Nortes X, et al. Mutant glialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am J Hum Genet. 2011;88:422–32. doi: 10.1016/j.ajhg.2011.02.009. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Lopez-Hernandez T, Sirisi S, Capdevila-Nortes X, Montolio M, et al. Molecular mechanisms of MLC1 and GLIALCAM mutations in megalencephalic leukoencephalopathy with subcortical cysts. Hum Molec Genet. 2011;20:3266–77. doi: 10.1093/hmg/ddr238. [PubMed] [CrossRef] [Google Scholar]
- Arnedo T, Lopez-Hernandez T, Jeworutzki E, Capdevila-Nortes X, Sirisi S, Pusch M, Estevez R. Functional analyses of mutations in HEPACAM causing megalencephalic leukoencephalopathy. Hum Mutat. 2014;35(10):1175–8. doi: 10.1002/humu.22622. [PubMed] [CrossRef] [Google Scholar]
- Mattina T, Perrotta CS, Grossfeld P. Jacobsen syndrome. Orphanet J Rare Dis. 2009;4:9. doi: 10.1186/1750-1172-4-9. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Grossfeld PD, Mattina T, Lai Z, et al. The 11q terminal deletion disorder: a prospective study of 110 cases. Am J Med Genet A. 2004;129A(1):51–61. doi: 10.1002/ajmg.a.30090. [PubMed] [CrossRef] [Google Scholar]
- Penny LA, Dell Aquila M, Jones MC, et al. Clinical and molecular characterization of patients with distal 11q deletions. Am J Hum Genet. 1995;56(3):676–83. [PMC free article] [PubMed] [Google Scholar]
- Guerin A, Stavropoulos DJ, Diab Y, et al. Interstitial deletion of 11q-implicating the KIRREL3 gene in the neurocognitive delay associated with Jacobsen syndrome. Am J Med Genet A. 2012;158A(10):2551–6. doi: 10.1002/ajmg.a.35621. [PubMed] [CrossRef] [Google Scholar]
- Ji T, Wu Y, Wang H, Wang J, Jiang Y. Diagnosis and fine mapping of a deletion in distal 11q in two Chinese patients with developmental delay. J Hum Genet. 2010;55(8):486–9. doi: 10.1038/jhg.2010.51. [PubMed] [CrossRef] [Google Scholar]
- Fryns JP, Kleczkowska A, Buttiens M, Marien P, van den Berghe H. Distal 11q monosomy. The typical 11q monosomy syndrome is due to deletion of subband 11q24.1. Clin Genet. 1986;30:255–60. doi: 10.1111/j.1399-0004.1986.tb00605.x. [PubMed] [CrossRef] [Google Scholar]
- Sirota L, Shabtai F, Landman I, et al. New anomalies found in the 11 q- syndrome. Clin Genet. 1984;26:569–73. doi: 10.1111/j.1399-0004.1984.tb01105.x. [PubMed] [CrossRef] [Google Scholar]
- Ono J, Hasegawa T, Sugama S, et al. Partial deletion of the long arm of chromosome 11: ten Japanese children. Clin Genet. 1996;50:474–8. doi: 10.1111/j.1399-0004.1996.tb02715.x. [PubMed] [CrossRef] [Google Scholar]
- Pivnick EK, Velagaleti GV, Wilroy RS, et al. Jacobsen syndrome: report of a patient with severe eye anomalies, growth hormone deficiency, and hypothyroidism associated with deletion 11 (q23q25) and review of 52 cases. J Med Genet. 1996;33(9):772–8. doi: 10.1136/jmg.33.9.772. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Wenger SL, Grossfeld PD, Siu BL, et al. Molecular characterization of an 11q interstitial deletion in a patient with the clinical features of Jacobsen syndrome. Am J Med Genet A. 2006;140(7):704–8. doi: 10.1002/ajmg.a.31146. [PubMed] [CrossRef] [Google Scholar]
- Tyson C, Qiao Y, Harvard C, Liu X, et al. Submicroscopic deletions of 11q24-25 in individuals without Jacobsen syndrome: re-examination of the critical region by high-resolution array-CGH. Mol Cytogenet. 2008;1:23. doi: 10.1186/1755-8166-1-23. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Van Zutven LJ, van Bever Y, Van Nieuwland CC, et al. Interstitial 11q deletion derived from a maternal ins(4;11)(p14;q24.2q25): a patient report and review. Am J Med Genet A. 2009;149A(7):1468–75. doi: 10.1002/ajmg.a.32714. [PubMed] [CrossRef] [Google Scholar]
- So J, Stockley T, Stavropoulos DJ. Periventricular nodular heterotopia and transverse limb reduction defect in a woman with interstitial 11q24 deletion in the Jacobsen syndrome region. Am J Med Genet A. 2014;164A(2):511–5. doi: 10.1002/ajmg.a.36292. [PubMed] [CrossRef] [Google Scholar]
- Yamamoto T, Shimada S, Shimojima K, et al. Leukoencephalopathy associated with 11q24 deletion involving the gene encoding hepatic and glial cell adhesion molecule in two patients. Eur J Med Genet. 2015 Sep;58(9):492–6. [PubMed]
- Khan A, Auger RR, Kushida CA, et al. Rhythmic movement disorder. Sleep Med. 2008 Mar;9(3):329–30. [PubMed]
- Takenouchi T, Hashida N, Torii C, et al. 1p34.3 deletion involving GRIK3: Further clinical implication of GRIK family glutamate receptors in thepathogenesis of developmental delay. Am J Med Genet A. 2014;164A(2):456–60. doi: 10.1002/ajmg.a.36240. [PubMed] [CrossRef] [Google Scholar]
- Pickard BS, Malloy MP, Christoforou A, et al. Cytogenetic and genetic evidence supports a role for the kainate-type glutamate receptor gene, GRIK4, in schizophrenia and bipolar disorder. Mol Psychiatry. 2006;11(9):847–57. doi: 10.1038/sj.mp.4001867. [PubMed] [CrossRef] [Google Scholar]
- Lopez-Hernandez T, Ridder MC, Montolio M, Capdevila-Nortes X, et al. Mutant glialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am J Hum Genet. 2011;88:422–32. doi: 10.1016/j.ajhg.2011.02.009. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Lopez-Hernandez T, Sirisi S, Capdevila-Nortes X, Montolio M, et al. Molecular mechanisms of MLC1 and GLIALCAM mutations in megalencephalic leukoencephalopathy with subcortical cysts. Hum Molec Genet. 2011;20:3266–77. doi: 10.1093/hmg/ddr238. [PubMed] [CrossRef] [Google Scholar]
- Arnedo T, Lopez-Hernandez T, Jeworutzki E, Capdevila-Nortes X, Sirisi S, Pusch M, Estevez R. Functional analyses of mutations in HEPACAM causing megalencephalic leukoencephalopathy. Hum Mutat. 2014;35(10):1175–8. doi: 10.1002/humu.22622. [PubMed] [CrossRef] [Google Scholar]
この記事の筆者:仲田洋美(医師)








