目次

📍 クイックナビゲーション
小児交互性片麻痺2型(AHC2)は、ATP1A3遺伝子の変異によって生じる、世界で約100万人に1人という超希少な神経発達疾患です。生後18か月未満に発症し、身体の左右が入れ替わるように繰り返す片麻痺発作、ジストニア、異常な眼球運動を特徴としますが、近年では「眠ると症状が消える」という独特な性質や、成人期にも進行する神経学的退行のリスクが明らかになり、生涯にわたる集学的医療が必要な疾患として再定義されつつあります。
Q. 小児交互性片麻痺2型(AHC2)とはどんな病気ですか?まず結論だけ知りたいです
A. ATP1A3遺伝子に新生突然変異(de novo変異)が生じることで発症する、生後18か月未満に始まる希少神経疾患です。身体の片側にエピソード性の麻痺が出現し、発作ごとに左右の症状が入れ替わる点が最大の特徴で、ジストニア・てんかん・発達遅滞などを伴います。睡眠で一時的に症状が消えるという独特な性質を持ちますが、根治療法はまだ確立されていません。
- ➤疾患の定義 → OMIM 614820、有病率約100万人に1人、AHC全体の約70〜80%を占める
- ➤分子メカニズム → ATP1A3変異によるNa⁺/K⁺ポンプ機能不全 → 皮質拡延性抑制(CSD)
- ➤主な症状 → 交互性片麻痺・ジストニア・てんかん(約50%)・自律神経症状・発達遅滞
- ➤遺伝子型と表現型 → D801N・E815K・G947Rの3大ホットスポット変異が約75%を占める
- ➤最新治療 → 高流量酸素療法の臨床試験とプライムエディティングによる遺伝子治療の進展
1. 小児交互性片麻痺2型(AHC2)とは:疾患の定義と歴史
小児交互性片麻痺(Alternating Hemiplegia of Childhood:AHC)は、生後18か月未満に発症する極めて稀かつ重篤な神経発達疾患です。1971年に独立した疾患概念として初めて医学文献に報告されて以来、その特異な臨床像から小児神経学における重要な研究対象となってきました。推定有病率は約100万人に1人とされています。
AHCは遺伝的に複数のサブタイプに分けられ、現在臨床的に確認されている症例の約70〜80%が、19番染色体長腕(19q13.2)に位置するATP1A3遺伝子のヘテロ接合性変異を原因とします。このタイプが「小児交互性片麻痺2型(AHC2、OMIM #614820)」として厳密に分類されています。残りの症例ではATP1A2遺伝子やRHOBTB2遺伝子の変異が原因となることも報告されており、遺伝的な異質性が知られています。
💡 用語解説:常染色体顕性(優性)遺伝とは
「常染色体」とは性染色体(X・Y)以外の染色体を指します。「顕性(優性)」とは、ペアになっている2本の染色体のうちどちらか1本に変異があるだけで症状が現れる遺伝形式です。AHC2はこの常染色体顕性(優性)遺伝形式をとりますが、両親から受け継がれる家族性の症例は非常に稀で、ほとんどは両親には変異がなく子どもで初めて生じる新生突然変異(de novo変異)によって発症します。
近年は、AHCが単一の疾患ではなく、ATP1A3遺伝子変異を基盤とする「ATP1A3関連疾患スペクトラム」という広範な表現型の一部として捉えられるようになっています。同じATP1A3遺伝子の変異であっても、変異の部位・種類によって急速発症型ジストニア・パーキンソニズム(RDP)やCAPOS症候群、てんかん性脳症など、まったく異なる疾患像を呈することが明らかになってきました。
2. 原因遺伝子ATP1A3と分子病態メカニズム
AHC2の原因であるATP1A3遺伝子は、神経細胞膜の電気的興奮性を支えるイオン輸送体「Na⁺/K⁺-ATPase(ナトリウム・カリウムポンプ)」のα3サブユニット(触媒部分)をコードしています。α3サブユニットは中枢神経系の大脳皮質・大脳基底核・海馬・小脳のニューロンや、心臓の刺激伝導系に高発現しています。

ATP1A3遺伝子がコードするα3サブユニットは、Na⁺/K⁺ポンプとして神経細胞内外のイオン勾配を維持する重要な役割を担います。
💡 用語解説:Na⁺/K⁺-ATPase(ナトリウム・カリウムポンプ)
細胞膜にあるタンパク質で、ATPのエネルギーを使って細胞内から3つのナトリウムイオン(Na⁺)を外に出し、細胞外から2つのカリウムイオン(K⁺)を内に入れるポンプの役割をします。この働きによって細胞膜を境にした電気的な勾配(静止膜電位)が保たれ、神経細胞は正常に電気信号を伝えられます。AHC2ではこのポンプの働きが弱まり、神経細胞が「過剰に興奮しやすい」状態になります。
変異 → ポンプ機能低下 → 皮質拡延性抑制(CSD)というカスケード
変異したα3サブユニットはイオン輸送能力が著しく低下し、細胞内外のイオン恒常性が崩壊します。神経細胞は静止膜電位を維持できず過剰な興奮状態(過興奮性)に陥ります。この細胞レベルの電気生理学的破綻が、AHC2における発作性運動障害・てんかん発作・持続的な発達遅滞を生み出す根本基盤になっています。
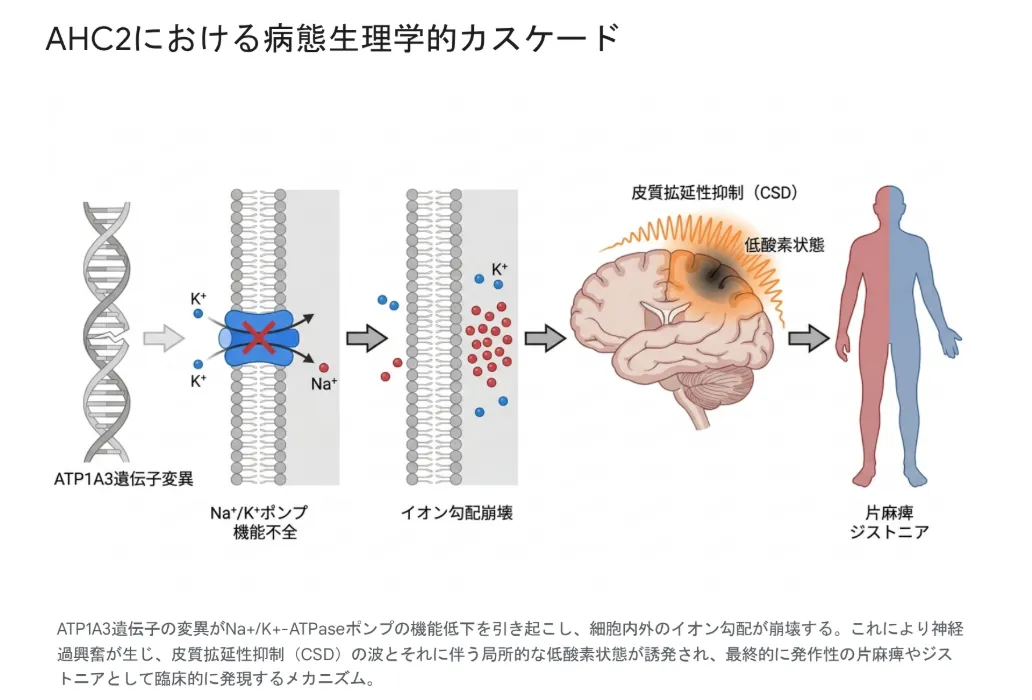
ATP1A3遺伝子変異 → Na⁺/K⁺ポンプ機能不全 → イオン勾配崩壊 → 皮質拡延性抑制(CSD)と局所的低酸素状態 → 片麻痺・ジストニアという病態生理的カスケード。
💡 用語解説:皮質拡延性抑制(CSD)
大脳皮質にゆっくりと広がっていく強力な電気的脱分極の「波」です。最初に神経細胞が急激に興奮した後、長時間の電気的沈黙(抑制)と局所的な低酸素状態を引き起こします。Na⁺/K⁺ポンプの機能不全はカリウムの細胞外クリアランスを遅らせるため、CSDの発生閾値が著しく下がります。後述する高流量酸素療法がAHC2の急性期発作に有効である可能性が示されているのは、このCSDに伴う低酸素状態を外部からの急速な酸素供給で和らげるという理論的根拠に基づいています。
3. 主な症状:発作性症状と持続性症状
AHC2の臨床像はきわめて複雑で、多彩な「発作性(エピソード性)症状」と、加齢とともに顕在化する「持続性(非発作性)症状」に大別されます。一つの疾患の中に複数の神経疾患が同居しているかのような症状を呈するため、患者さんとご家族の生活への影響は甚大です。
代表的な発作性症状
👁 異常眼球運動
単眼性または両眼性の眼振、斜視、共同偏視、眼球ボビングなど。多くの場合、疾患の最初の兆候として現れ、ほかの運動症状に先行することが多い。
🦵 片麻痺・四肢麻痺
突然の筋力低下が身体の片側に出現し、発作ごとに左右の罹患側が交代することが最大の特徴。重症例では両側性の四肢麻痺に移行することもある。
💪 ジストニアと強直発作
痛みを伴う持続的な異常な筋収縮や姿勢異常が、単独または片麻痺と混在して出現。全身性ジストニア発作を呈することもある。
⚡ てんかん発作
患者さんの最大約50%が経過中にてんかんを合併。片麻痺やジストニアとは病態生理が異なり、従来の抗てんかん薬への反応が不十分で難治化することがある。
💡 用語解説:ジストニアと片麻痺
ジストニアとは、自分の意志とは関係なく筋肉が持続的に収縮し、ねじれを伴う異常な姿勢や反復運動が生じる状態。痛みを伴うことがあります。片麻痺は身体の左右どちらかの手足や顔面に筋力低下が起こる状態で、AHC2では数分〜数日、まれに数週間続くこともあります。
発作の誘因と「睡眠で症状が消える」現象
AHC2の発作はランダムに起こるだけでなく、特定の誘因で誘発されることが知られています。精神的ストレス・興奮・極端な温度変化(入浴や冷水・温水)・睡眠不足・空腹・強い蛍光灯の光などが主な誘発因子です。日常生活で誘因を避ける工夫が発作予防の第一歩になります。
そして、AHC2の最も特異な臨床的特徴の一つが「発作中に眠ると、片麻痺やジストニアの症状が一時的に、あるいは完全に消失する」という現象です。発作の最中に眠りに落ちると睡眠中は正常な筋緊張に戻り、覚醒後数分〜数十分以内に再び症状が出現するケースが多くみられます。この「睡眠によるリセット」は古くから診断的特徴とされてきましたが、近年のポリソムノグラフィ研究でAHC2患者さんの睡眠そのものが完全には正常ではないことも明らかになっており、夜間てんかんや中枢性睡眠時無呼吸への注意が必要です。
持続性症状と全身合併症
年齢を重ねるにつれて、一過性の発作頻度は変化(一部の症例では減少)する一方で、持続的かつ非発作性の神経機能障害が前景に立つようになります。ほぼ全ての患者さんが軽度から重度の知的障害および精神運動発達遅滞を呈し、加齢とともに認知機能が低下していくケースが多く報告されています。
運動面では小脳失調・構音障害・舞踏アテトーゼ・パーキンソニズム・持続的ジストニアが定着し、歩行や巧緻運動に重大な影響をもたらします。さらにATP1A3は心臓にも発現するため、心室性不整脈などの心伝導系異常を発症するリスクが高く、心機能の定期的なモニタリングが必須です。麻酔や鎮静時の合併症リスク(徐脈・無呼吸)も高く、外科手術の際には特別な配慮が必要です。

4. ATP1A3関連疾患スペクトラムと遺伝子型・表現型相関
ATP1A3遺伝子の変異はAHC2という単一の疾患だけでなく、さまざまな神経疾患を引き起こすことが明らかになっています。これらは現在、「ATP1A3関連疾患(ATP1A3-related disorders)」という包括的なスペクトラムとして捉えられ、明確な境界を持たず、一人の患者さんで複数の表現型が重なり合うこともあります。
💡 ATP1A3関連疾患スペクトラムの代表例
- ➤小児交互性片麻痺2型(AHC2):生後18か月までに発症する交互性片麻痺・ジストニア
- ➤急速発症型ジストニア・パーキンソニズム(RDP/ジストニア12型):生後18か月から成人期まで幅広い発症年齢。感染症・出産・ストレスを契機に急速発症
- ➤CAPOS症候群:小脳失調・無反射・凹足・視神経萎縮・感音難聴の頭文字。発熱を契機に乳幼児期に急性発症
- ➤RECA / FIPWE:発熱・ウイルス感染を契機に小脳失調や脳症エピソードを反復
- ➤早期乳児てんかん性脳症(DEE-99など):難治性てんかんと重度発達遅滞を呈する
3大ホットスポット変異と重症度の関係
ATP1A3には現在までに100種類以上の病的変異が同定されていますが、AHC2患者さんの約75%は3つのホットスポット変異(D801N、E815K、G947R)のいずれかを有することが、大規模コホート研究で判明しています。変異の種類によって臨床的重症度・予後・薬剤反応性が明確に異なります。
AHC2症例における主要なATP1A3変異の頻度分布
36.5%
25.5%
11.5%
D801N(軽症傾向・自立歩行獲得率が高い)、E815K(早期発症・重度ジストニア・自立困難)、G947R(中間的重症度)の3変異でAHC2症例の約75%を占めます。データはイタリアおよび国際コホート研究に基づきます。
特にE815K変異は発症年齢が早く、重度の知的障害と運動機能低下を伴う最も重篤な表現型を呈します。フルナリジンへの反応性は比較的良好ですが、ベースラインの神経学的障害が重度のため長期予後は不良です。一方D801N変異は発症がやや遅く、運動機能の障害は中等度にとどまり、自立歩行を獲得する患者さんが多い傾向があります。
5. 診断基準と遺伝子検査の進め方
AHC2の診断は、遺伝子検査が普及した現在でも詳細な病歴聴取と特異的臨床症状の観察に基づく臨床診断が基本です。1993年にAicardiらが提唱し、2015年にPanagiotakakiらによって更新された国際的な診断基準が現在も広く用いられています。
💡 AHC国際診断基準(要約)
- ➤生後18か月未満における発作性イベントの発症
- ➤左右いずれかの半身を交互に巻き込む反復性片麻痺発作
- ➤片麻痺発作の汎化、または両側性に発症する四肢麻痺エピソード
- ➤強直・ジストニア発作、眼振、斜視、自律神経症状など他の発作性異常の併存
- ➤精神運動発達遅滞や持続的神経学的異常の存在
- ➤発作性症状が睡眠によって少なくとも一時的に消失すること
急性期の鑑別と画像診断
脳MRIは通常、特異的な異常を示さず正常か、一部の症例で局所的な脳萎縮や軽度の小脳萎縮を認める程度です。そのため急性期に突然の片麻痺で救急搬送された乳幼児では、小児脳卒中(動脈虚血性脳卒中)・急性散在性脳脊髄炎(ADEM)・もやもや病など生命を脅かす他疾患を迅速に除外することが救急管理上きわめて重要となります。
確定診断はトリオ全エクソーム解析(Trio-WES)が第一選択
💡 用語解説:トリオ全エクソーム解析(Trio-WES)
遺伝子のうちタンパク質をコードしている領域(エクソン)を網羅的に解析する次世代シーケンスを、患者さんご本人と両親3名で同時に行う方法です。両親には変異がなく子どもで初めて生じた新生突然変異(de novo変異)を効率よく検出できるため、AHC2のように多くが新生突然変異で発症する疾患の確定診断にきわめて有効です。
表現型スペクトラムが広いため、ATP1A3単一遺伝子検査では情報が十分でないことが多く、ATP1A2やRHOBTB2など類縁遺伝子も含めた包括的なパネル検査または全エクソーム解析が推奨されます。当院で提供している関連検査としては以下のパネルにATP1A3が含まれています。
- ➤ジストニア・ジスキネジアNGSパネル検査(25遺伝子) — AHC2およびRDPを含む遺伝性ジストニア・ジスキネジアの主要原因遺伝子をカバー
- ➤てんかん包括的NGSパネル検査(1057遺伝子) — 新生児期から成人期まで、てんかん性脳症を含むあらゆるてんかんの原因遺伝子を網羅
- ➤NIPTインペリアルプラン(154遺伝子218疾患) — 出生前にATP1A3を含む単一遺伝子疾患をスクリーニング
6. 治療戦略と最新研究の最前線
AHC2およびATP1A3関連疾患に対する根治的な治療法は現時点ではまだ確立されていません。治療方針は、発作の予防・急性発作の迅速な管理・多面的な合併症への対症療法とリハビリテーションからなる集学的アプローチ(小児神経・循環器・精神科・耳鼻咽喉科・リハビリテーションなどとの連携)が基本です。
発作予防の中心:フルナリジン
薬物療法において世界的にもっとも広く使用されているのが、カルシウム・ナトリウムチャネル阻害薬であるフルナリジン(Flunarizine)です。神経細胞の異常な過興奮を抑え、皮質拡延性抑制(CSD)の発生閾値を上昇させることで発作の頻度と強度を軽減すると考えられています。特に重症型E815K変異の患者さんで発作抑制効果が高いことが示唆されているほか、長期にわたる継続投与が発作による興奮毒性から神経細胞を保護し、不可逆的な神経学的退行の予防にも寄与する可能性が報告されています。
てんかんを合併する場合はそのタイプに合わせた抗てんかん薬を併用し、CSDの抑制を目的としたケトン食療法の臨床応用も検討されています。日常生活では、発作の誘因となる極端な温度変化・強い光・過度のストレス・睡眠不足の回避が重要です。
急性期治療の新しい選択肢:高流量酸素療法
パリ脳研究所を中心とする研究グループから、急性発作時に100%医療用酸素を12 L/分という高流量で投与することで、片麻痺やジストニアの発作を劇的に沈静化できる可能性が報告されています。この治療は皮質拡延性抑制(CSD)に伴う局所的低酸素状態を急速な酸素供給で和らげるという理論的根拠に基づいており、現在第2相臨床試験(NCT06248645)が進行中で、発作開始後30分以内の症状停止割合を主要評価項目として有効性が検証されつつあります。
遺伝子治療の未来:プライムエディティングによる根治の可能性
💡 用語解説:プライムエディティング(Prime Editing)
CRISPR由来の精密な遺伝子編集技術で、DNAの二本鎖を切断せずに目的の塩基配列だけをピンポイントで書き換えることができます。従来のCRISPR/Cas9で問題となっていた予期せぬオフターゲット変異や挿入・欠失のリスクを大幅に下げられるのが特長です。
マサチューセッツ工科大学・ハーバード大学Broad Instituteの研究チームは、AHC2マウスモデルに対しAAV9ベクターを用いたプライムエディティングを適用し、大脳皮質で最大48%のDNA修正と73%のmRNA修正を達成、寿命を2倍以上に延長することに成功しました。低体温誘発性発作・運動機能・認知機能のすべてで顕著な改善が確認されており、これは「症状を和らげる対症療法」から「DNAレベルで疾患の進行を止める疾患修飾療法」へのパラダイムシフトを示す画期的な成果です。今後ヒト臨床応用に向けた研究が進められていますが、安全性・送達手法・最適な治療時期など、克服すべき課題も残されています。
7. 遺伝カウンセリングの意義
AHC2の確定診断がついた後、ご家族への丁寧な遺伝カウンセリングが欠かせません。臨床遺伝専門医が中心となり、以下の内容を扱います。
- ➤再発リスクの説明:AHC2のほとんどは新生突然変異(de novo変異)であり、両親には変異が確認されません。ただし生殖細胞モザイクの可能性は完全には除外できないため、次子の妊娠時には出生前診断の選択肢が議論の対象になります。患者さんご本人が将来子どもを持つ場合、遺伝確率は理論上50%です。
- ➤出生前診断の選択肢:家族内で変異が同定されている場合、絨毛検査・羊水検査による出生前遺伝子診断が可能です。ただし表現型のばらつきが大きく、検査を受けるかどうかはご家族の価値観に基づいて慎重に判断する必要があります。
- ➤長期的な医療計画:心伝導系異常・突然死リスク・成人期の退行リスクを念頭に、循環器・神経・精神科などとの継続的な連携体制を構築します。
- ➤心理社会的サポート:希少疾患ゆえに情報が限られ、ご家族の孤立感が深まりやすい疾患です。患者団体や同じ疾患を持つご家族との連携、教育・福祉資源へのアクセスを支援します。
8. よくある誤解
誤解①「眠れば治るから心配ない」
睡眠で症状が一時的に消失するのはAHC2の特異な性質ですが、根本的な治癒ではありません。夜間てんかん発作や中枢性睡眠時無呼吸が隠れていることがあり、突然死のリスク要因にもなります。
誤解②「進行しない静的な病気」
かつてはそう考えられていましたが、成人期の長期追跡研究で不可逆的な神経学的退行や若年での突然死リスクが明らかになっています。生涯にわたる集学的医療が必要です。
誤解③「片麻痺=脳卒中だから別の病気」
乳幼児で繰り返す片麻痺の多くは脳卒中ではなく、AHCを含む遺伝性疾患の可能性があります。脳MRIが正常でも臨床診断は可能であり、安易な脳卒中診断で遺伝子検査が遅れることが課題です。
誤解④「親に同じ症状がないから遺伝ではない」
AHC2の大多数は新生突然変異(de novo変異)で発症します。両親が健康でも、お子さんで初めて遺伝子に変化が生じることは珍しくありません。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 希少神経疾患の診断・遺伝カウンセリングについて
AHC2をはじめとするATP1A3関連疾患のご相談は、
臨床遺伝専門医が常駐するミネルバクリニックへお気軽にご相談ください。
関連記事
参考文献
- [1] Brashear A, Sweadner KJ, Cook JF, et al. ATP1A3-Related Disorder. GeneReviews®. NCBI Bookshelf. [NCBI Bookshelf]
- [2] OMIM #614820. Alternating Hemiplegia of Childhood 2; AHC2. Johns Hopkins University. [OMIM]
- [3] Heinzen EL, Swoboda KJ, Hitomi Y, et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat Genet. 2012;44(9):1030-1034. [PMC9351909]
- [4] Vezyroglou A, Varadkar S, Bast T, et al. Alternating Hemiplegia of Childhood: Genotype–Phenotype Correlations in a Cohort of 39 Italian Patients. Front Neurol. 2021;12:658451. [Frontiers]
- [5] Panagiotakaki E, De Grandis E, Stagnaro M, et al. Evidence of a non-progressive course of alternating hemiplegia of childhood. Brain. 2010;133(12):3598-3610. [Brain]
- [6] Uchitel J, Helseth A, Prange L, et al. Non-Stationary Outcome of Alternating Hemiplegia of Childhood into Adulthood. Ann Clin Transl Neurol. 2022. [PMC8810436]
- [7] Paris Brain Institute. Oxygen therapy subsides abnormal movements in a rare childhood disease. [Paris Brain Institute]
- [8] ClinicalTrials.gov. Oxygen As an Acute Treatment in Alternating Hemiplegia of Childhood (NCT06248645). [ClinicalTrials.gov]
- [9] Broad Institute. Prime editing treats childhood brain disease in mice. [Broad Institute]
- [10] Sasaki M, Sumitomo N, Shimizu-Motohashi Y, et al. Alternating Hemiplegia of Childhood and ATP1A3-Related Diseases. Neurol Genet. 2024. [Neurol Genet]






