目次

📍 クイックナビゲーション
FGFR3(線維芽細胞増殖因子受容体3)は、骨格発生においては軟骨細胞の増殖を強力に抑制する「ブレーキ」として、上皮組織においては細胞の異常増殖を促進する「がん遺伝子(アクセル)」として作用するという、生物学的に極めて特異な二面性を持つ受容体チロシンキナーゼです。生殖細胞系列の機能獲得型変異は軟骨無形成症(出生26,000〜28,000人に1人)をはじめとする先天性骨系統疾患を引き起こし、体細胞変異は膀胱がん・多発性骨髄腫・良性皮膚腫瘍の発症ドライバーとなります。近年、ボソリチドやインフィグラチニブなど画期的な分子標的治療が相次いで承認・開発されており、精密医療の最前線領域として大きな注目を集めています。
Q. FGFR3遺伝子とはどのような遺伝子ですか?まず結論だけ知りたいです
A. 細胞増殖・分化・骨格発生を制御する受容体チロシンキナーゼをコードする遺伝子です。骨組織では軟骨細胞の増殖を「抑制」し、上皮組織では増殖を「促進」するという相反する二面性が最大の特徴です。変異の種類(生殖細胞系列 vs 体細胞)と発現組織の組み合わせによって、先天性骨疾患にもがんにもなりうる、遺伝学上きわめて重要な遺伝子です。
- ➤遺伝子の基本 → 4番染色体短腕(4p16.3)に位置する受容体チロシンキナーゼファミリーの一員
- ➤シグナル伝達 → MAPK/ERK経路・STAT1経路を介して骨成長を負に制御、Snail1フィードバックで増幅
- ➤先天性骨系統疾患 → 軟骨無形成症(ACH)・致死性骨異形成症(TD)・ミュンケ症候群など7疾患スペクトラム
- ➤がん・腫瘍 → 膀胱がん(NMIBC 39%)・多発性骨髄腫(全体の約15%)・良性皮膚腫瘍(脂漏性角化症の39%)
- ➤最新治療 → ボソリチド(承認済み)・インフィグラチニブ(第III相完了)・ダボグラチニブ(開発中)
1. FGFR3遺伝子とは:概要と生体内での役割
FGFR3(Fibroblast Growth Factor Receptor 3:線維芽細胞増殖因子受容体3)は、細胞の増殖・分化・血管新生・創傷治癒・胚発生などの極めて重要な細胞プロセスを制御する、高度に保存された膜貫通型タンパク質をコードする遺伝子です。FGFR1〜4からなる受容体チロシンキナーゼ(RTK)ファミリーの一員であり、特定の線維芽細胞増殖因子(FGF)リガンドと結合して細胞内にシグナルを伝達します。FGFR3遺伝子は4番染色体短腕(4p16.3)に位置し、進化的にきわめて高度に保存されています(ヒトとマウスでアミノ酸配列の約92%が一致)。この高い保存性は、FGFR3が生命の発生・維持において根本的かつ不可欠な機能を担っていることを示しています。
💡 用語解説:受容体チロシンキナーゼ(RTK)とは
受容体チロシンキナーゼ(Receptor Tyrosine Kinase)とは、細胞の外側でリガンド(信号分子)を受け取り、細胞内でチロシンというアミノ酸をリン酸化することでシグナルを伝える膜貫通型タンパク質の一群です。FGFR3はこのRTKファミリーに属し、細胞外ドメイン・膜貫通ドメイン・細胞内チロシンキナーゼドメインの3つから構成されています。がんや先天性疾患の主要な標的分子として、多くの分子標的薬開発の中心となっています。
生体においてFGFR3は、組織のコンテクストに依存してまったく相反する二面性を示します。骨組織の発生においては、軟骨細胞の増殖と分化を強力に抑制することで骨の過剰な伸長を防ぐ「ブレーキ」として機能します。一方で、膀胱などの上皮組織においては、細胞の異常増殖を促進し発がんを駆動する「アクセル(がん遺伝子)」として作用します。この組織特異的な機能の相違は、下流のシグナル伝達経路の複雑なクロストークと、各組織における遺伝子発現プロファイルの違いによって説明されます。
💡 用語解説:スプライシングバリアント(アイソフォーム)とは
ひとつの遺伝子から、mRNAの一部を切り取り・繋ぎ合わせる「選択的スプライシング」によって複数の異なるタンパク質が作られることがあります。これらをスプライシングバリアント(アイソフォーム)といいます。FGFR3では骨を形成する細胞群と上皮細胞群で異なるアイソフォームが発現しており、これが同じFGFR3遺伝子産物でありながら組織によってまったく異なる機能(増殖の抑制 vs 促進)をもたらす根本的な理由となっています。
FGFR3の機能の重要性は「逆方向の証拠」からも裏付けられています。機能喪失型変異(Loss-of-Function)によって生じるCATSHL症候群(屈指症・高身長・側弯症・難聴を伴う)では、骨が過剰に成長するという正反対の表現型が出現します。これは生体においてFGFR3が本来「骨成長の負の調節因子」として不可欠な役割を果たしていることを分子遺伝学的に明確に示しています。
2. 分子構造とシグナル伝達経路の全容
受容体の基本構造とリガンド結合
FGFR3タンパク質は細胞膜を貫通する構造を持ち、主に3つの機能ドメインから構成されています。①細胞外リガンド結合ドメイン(酸性・塩基性FGFと特異的に結合)、②単一の膜貫通ドメイン、③細胞内チロシンキナーゼドメインです。内軟骨性骨化のプロセスにおいては、軟骨膜および周囲の組織から分泌されるFGF9およびFGF18が主要なパラクリンリガンドとして機能します。これらのリガンドが細胞外マトリックス内のヘパラン硫酸(HS)と複合体を形成しながらFGFR3に結合することで、受容体の二量体化が誘導され、細胞内ドメインの自己リン酸化が引き起こされます。
FGFR3シグナル伝達経路と軟骨形成の阻害機構
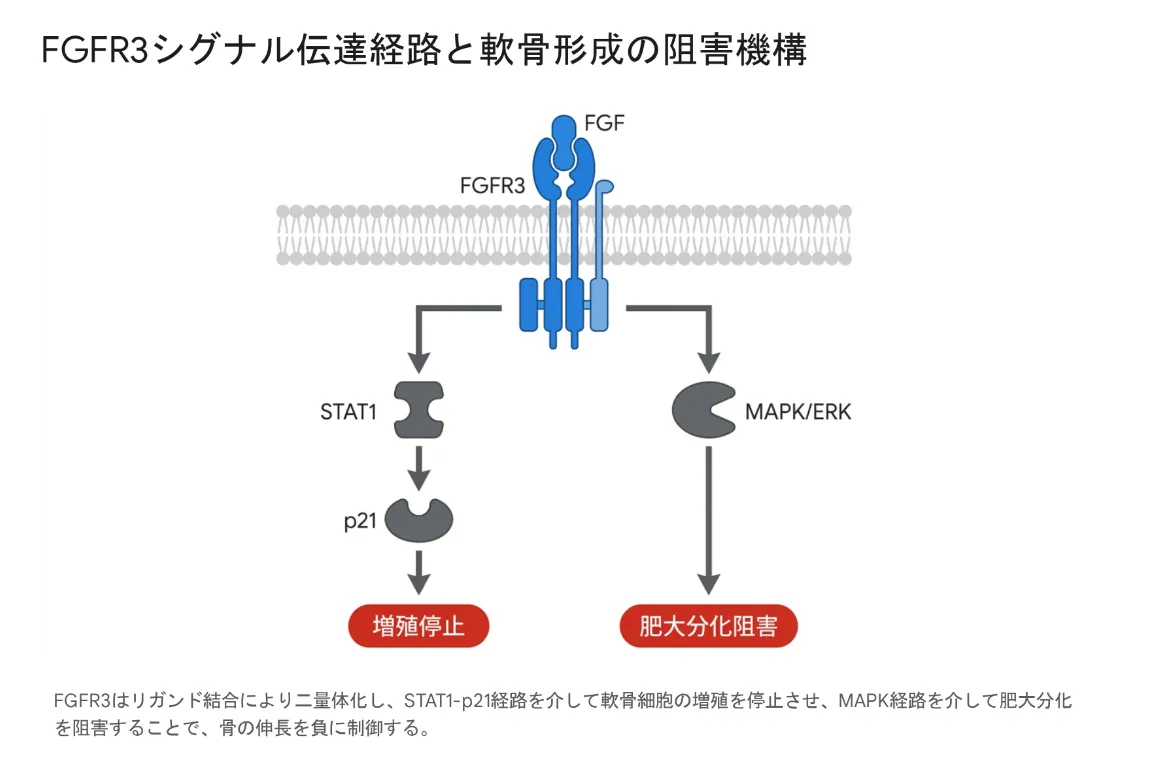
主要シグナル経路①:MAPK(RAS-RAF-MEK-ERK)経路
💡 用語解説:MAPK経路(RAS-RAF-MEK-ERK)とは
MAPK(分裂促進因子活性化タンパク質キナーゼ)経路は、RAS→RAF→MEK→ERKというキナーゼの順次活性化を伴う細胞内シグナル伝達の主要経路です。最終的にERK1/2が活性化されると核内に移行して遺伝子発現を変化させます。FGFR3の活性化によってこの経路が駆動されると、軟骨細胞の増殖停止と分化制御が引き起こされます。がん細胞ではこの経路が恒常的に活性化されることで細胞増殖が促進されます。
FGFR3の活性化はERK1/2のリン酸化を促進し、細胞核内での遺伝子発現をダイナミックに変化させます。興味深い知見として、生体内での長期間の機械的ストレスはERK1/2のリン酸化を抑制する一方で、骨芽細胞様細胞株(MC3T3-E1)においてはFGFR3受容体の発現そのものを増加させるという代償的なフィードバック機構の存在が示されています。また、構成的に活性化されたMEK1を発現させると、FGFR3欠損マウスに見られる骨の過成長を軟骨細胞の分化阻害によって「レスキュー(救済)」できることが確認されており、MAPK経路が軟骨細胞の分化制御に必須であることが証明されています。
主要シグナル経路②:STAT1経路とSnail1フィードバックループ
💡 用語解説:STAT1経路とは
STAT1(シグナル伝達兼転写活性化因子1)は、リン酸化されると核内に移行して標的遺伝子の転写を直接活性化する転写因子です。FGFR3が活性化されるとSTAT1がリン酸化・核内移行し、細胞周期を止める阻害因子p21(p21Waf1/Cip1)の発現を強力に誘導します。このSTAT1-p21経路によって成長板軟骨細胞の増殖が停止し、一方でMAPK/ERK経路は軟骨細胞の肥大分化を阻害します(上図の二分岐に対応)。致死性骨異形成症(Thanatophoric Dysplasia)の骨組織では、下流の転写因子Snail1が極めて高いレベルで異常発現していることも確認されています。
FGFR3が活性化されると、MAPK経路・STAT1経路の双方によって転写因子Snail1の発現が強力に誘導されます。Snail1はさらにSTAT1とMAPK経路の両方を活性化するという正のフィードバックループを形成することで、FGFR3シグナルを自己増幅させます。この連鎖によってp107・p21Waf1/Cip1・Sox9などの下流シグナル分子が活性化され、成長板軟骨細胞の増殖および肥大軟骨細胞への分化が抑制されます。結果として「早期老化(Premature Senescence)」と呼ばれる特異的な増殖停止状態が引き起こされ、内軟骨性骨化が強力にブレーキをかけられます。
さらに、成長板におけるFGFRシグナルの微調整には、Nf1遺伝子がコードするニューロフィブロミン(Ras-GAP:GTPase活性化タンパク質)が関与しています。ニューロフィブロミンは前肥大軟骨細胞においてFGFR1およびFGFR3と共発現しており、Ras-ERKシグナルを抑制することで、破骨細胞形成を促進するFGFR1シグナルを減弱させるという複雑な細胞内制御機構が存在しています。
組織特異的アイソフォームが生み出す「正反対の表現型」
代替スプライシングによって生み出される複数のアイソフォームが、FGFR3の組織特異的な機能の鍵を握っています。骨を形成する細胞群(軟骨細胞・骨芽細胞)には増殖を「抑制」する方向のアイソフォームが発現する一方、皮膚の表皮細胞を含む上皮細胞群にはまったく異なる特定のアイソフォームが発現します。このコンテクスト依存的な切り替えにより、同一遺伝子から生み出される受容体であっても、骨組織では「増殖の強力なブレーキ」として、上皮細胞・がん細胞では「増殖の強力なアクセル」として機能するという相反する表現型がもたらされます。
3. FGFR3遺伝子変異に起因する先天性骨系統疾患
💡 用語解説:機能獲得型変異(Gain-of-Function Mutation)とは
機能獲得型変異(GoF変異)とは、変異によってタンパク質が本来のリガンド(信号分子)がなくても常時「オン」の活性化状態になってしまう変異です。FGFR3のGoF変異では、FGF9やFGF18が結合していなくても受容体が恒常的にシグナルを発し続けるため、骨成長の「ブレーキ」が踏みっぱなしになります。その結果、軟骨細胞の増殖・分化が過剰に抑制され、骨の長軸成長が深刻に障害されます。
💡 用語解説:生殖細胞系列変異 vs 体細胞突然変異
生殖細胞系列変異とは、精子・卵子などの生殖細胞に生じた変異で、受精によって生まれたすべての細胞に引き継がれます。FGFR3の骨系統疾患はこちらが原因です。一方体細胞突然変異とは、生後に特定の組織・細胞で新たに生じた変異で、その個体の子どもには遺伝しません。FGFR3が関与する膀胱がんや脂漏性角化症はこちらが原因です。同じFGFR3変異でも、どちらの細胞で起きたか・どの組織で起きたかによって、まったく異なる疾患を引き起こすことが重要なポイントです。
軟骨無形成症(Achondroplasia:ACH)
軟骨無形成症(ACH)は、不均衡な低身長を特徴とする最も一般的な骨系統疾患であり、出生26,000〜28,000人に1人の割合で発生します。常染色体優性遺伝形式をとりますが、影響を受ける個人の約80%は平均的な身長の両親から生まれ、これらは生殖細胞系列での「新規(de novo)」の病原性バリアントに起因します。
💡 用語解説:ホットスポット変異とは
ホットスポット変異とは、特定の遺伝子の特定の位置に変異が異常に高頻度で集中する現象です。ACHの場合、推定99%の症例でFGFR3遺伝子の1138番目の塩基(c.1138G>A または c.1138G>C)というひとつの位置に変異が集中します。このような極端な局在性は自然界では非常に珍しく、この部位が変異しやすい配列的な特性(CpGジヌクレオチドでの脱アミノ化)によると考えられています。父親の加齢とともにde novo変異の頻度が上昇することも知られています。
ほぼすべての症例(推定99%)で、FGFR3遺伝子の特定のホットスポット変異(c.1138G>A または c.1138G>C)が認められます。これはタンパク質の380番目のグリシンがアルギニンに置換される(p.Gly380Arg)結果をもたらし、細胞膜上のFGFR3受容体が過剰にシグナルを生成し、正常な骨の伸長を打ち消します。ACH患者同士の妊娠においては、50%の確率でACHの子どもが生まれるほか、25%の確率でホモ接合体軟骨無形成症(重篤な胸郭低形成を伴う致死的状態)となるリスクがある点も重要です。
臨床的特徴として、患者は近位(根部)の四肢短縮・大頭症・前頭部の突出(Frontal bossing)・鼻根部の陥凹を伴う顔面中央部の低形成を呈します。新生児期には狭い胸郭や「三叉槍(trident)」状の指の配置が見られ、乳児期には筋緊張低下が頻繁に観察されます。発達上の運動マイルストーンの獲得は遅れる傾向がありますが、知的発達は通常正常範囲です。
医学的合併症は多岐にわたります。閉塞性睡眠時無呼吸・頻繁な中耳機能障害・O脚(内反膝)・胸腰椎の後弯症・成人期の脊柱管狭窄症などに対して継続的な管理が必要です。特に乳児期の大後頭孔狭窄による頭蓋頸椎移行部の圧迫は死亡リスクを増加させるため、生後6〜12ヶ月ごとの厳密な神経学的・画像的モニタリングが必須であり、下肢反射亢進・クローヌス・睡眠ポリグラフでの中枢性無呼吸増加などが確認された場合は後頭下減圧術などの外科的介入が適応となります。日常生活においては、頭蓋頸椎への衝撃を避けるためトランポリン・衝突を伴うスポーツ・自動ベビースイングの使用を避けるべきであり、妊婦においては骨盤が著しく狭小であるため帝王切開での分娩が必須とされます。
FGFR3関連疾患スペクトラム:変異の強さが重症度を決める
FGFR3遺伝子の異なる部位におけるバリアントは、変異の強さと細胞への影響度合いに応じて、ACH以外にも多様なスペクトラムの疾患を引き起こします。
| 疾患名 | 重症度 | 主な臨床的特徴 | 代表的な遺伝子変異 |
|---|---|---|---|
| 軟骨無形成症(ACH) | 中等度〜重度 | 不均衡な低身長、根部短縮、大頭症、脊柱管狭窄、頭蓋頸椎圧迫 | p.Gly380Arg(c.1138G>A/C) |
| 軟骨低形成症(HCH) | 軽度 | ACHより軽度の骨格異常、知的障害・てんかんのリスクあり、黒色表皮腫の合併例も | p.Asn540Lys など |
| 致死性骨異形成症(TD) I型 / II型 |
致死的 | 極端な四肢短縮、重度の胸郭低形成、新生児期の呼吸不全で通常死亡 | Y373C、K650E など |
| SADDAN症候群 | 極めて重度 | 重度の低身長、重度の発達遅延、黒色表皮腫(皮膚の色素沈着)を伴う | K650M |
| ミュンケ症候群 | 部位特異的 | 冠状縫合の早期癒合、大頭、難聴、手足の異常 | P250R |
| 黒色表皮腫伴発型 クルーゾン症候群 |
部位特異的 | 頭蓋骨縫合の早期癒合、顔面の変形、皮膚のしわ・ひだの厚いビロード状色素沈着(黒色表皮腫) | A391E(p.Ala391Glu) |
| LADD症候群 | 軽度〜中等度 | 涙液・唾液の産生低下、耳の奇形と難聴、小さな歯、手の変形 | D513N(p.Asp513Asn) |
HCHではACHと類似の骨格異常が軽度に見られますが、特筆すべきは知的障害やてんかんの発生率がACHより高い場合があること、さらに稀にインスリン抵抗性と二次性高インスリン血症を伴う黒色表皮腫(Acanthosis nigricans)がHCH患者で報告されており、上皮細胞におけるFGFR3機能変容を示す新たな臨床的関連性が示唆されています。ミュンケ症候群のP250R変異は骨の長軸成長ではなく頭蓋骨の縫合線の早期癒合を引き起こす点で他の骨系統疾患とは異なる表現型を示します。同様に頭蓋骨に影響する黒色表皮腫伴発型クルーゾン症候群(A391E変異)は、頭蓋骨縫合の早期癒合に加え、皮膚のしわやひだに厚いビロード状の色素沈着(黒色表皮腫)を伴います。またLADD症候群(D513N変異)は涙液・唾液の産生低下、耳の奇形と難聴、小さな歯、手の変形を特徴とし、FGFR3変異が長管骨の成長だけでなく頭蓋・外胚葉系組織の発生にも広く関与していることを示しています。
機能喪失型変異が引き起こす「逆方向」の疾患:CATSHL症候群
💡 用語解説:機能喪失型変異(Loss-of-Function)とは
機能喪失型変異(LoF変異)とは、変異によってタンパク質本来のはたらきが低下・消失する変異です。FGFR3は骨成長の「ブレーキ」なので、このブレーキが弱まると骨の成長が止まらず、機能獲得型変異(骨が縮む)とは正反対に骨が過剰に伸びる表現型が現れます。同じ遺伝子でも、変異が「強める」方向か「弱める」方向かで真逆の病気になる——これがFGFR3の二面性をもう一つの角度から示しています。
その代表例がCATSHL症候群(Camptodactyly, Tall stature, Scoliosis and Hearing Loss syndrome)です。病名のとおり屈指症(指の屈曲拘縮)・高身長・側弯症・難聴を主徴とし、常染色体優性遺伝の形式をとります。ある家系では7世代・約30人にわたって受け継がれ、完全な浸透率(変異を持つ人はほぼ全員発症)を示すことが確認されています。原因はFGFR3のミスセンス変異(N540S、R621H、新規に報告されたR621Cなど)で、骨成長を抑える働きが部分的に失われることで発症します。R621C変異の症例では、通常CATSHLには見られないLADD症候群に典型的な円錐歯(peg-shaped incisors)が観察されており、FGFR3関連疾患どうしの表現型の重複も示唆されています。
病態メカニズムの解明には、FGFR3を欠損させたゼブラフィッシュモデルが用いられています。このモデルでは小頭症や頭蓋骨縫合の閉鎖遅延、軟骨腫様の病変、聴覚器官の発達異常など、ヒトのCATSHL患者に似た表現型が再現されます。分子レベルでは、FGFR3の機能喪失によってインディアンヘッジホッグ(IHH)シグナルとWnt/β-カテニンシグナルがともに亢進していることが判明しました。注目すべきことに、Wnt/β-カテニン活性を薬理学的に阻害すると変異ゼブラフィッシュの表現型が部分的に改善することが示されており、この経路がCATSHL症候群や関連する骨格疾患の将来的な治療標的になりうる可能性を示しています。

4. 体細胞突然変異とがん・腫瘍形成
生殖細胞系列で骨格形成異常を引き起こすのとまったく同一のFGFR3活性化変異が、成長後の生体組織(体細胞)において発生した場合、それは発がんプロセスを駆動する強力な「がん遺伝子(オンコジーン)」として作用します。これは細胞特異的な下流経路(骨では増殖停止に働くメカニズムが、上皮では欠如または修飾されていること)が変異の影響を反転させるためであり、発現組織によって正反対の病理学的結果をもたらす典型例です。
💡 用語解説:がん遺伝子(オンコジーン)とは
がん遺伝子(oncogene)とは、変異・増幅・過剰発現などによって細胞増殖や生存シグナルを過剰に活性化し、がんの発生・進展を促進する遺伝子です。正常状態では「プロトオンコジーン(がん原遺伝子)」として適切に調節されていますが、ひとつのアレルに変異が生じるだけで(優性的に)がん化を促進しうる点が、がん抑制遺伝子(両方のアレルが変異して初めて機能喪失する)と異なります。FGFR3は上皮組織ではオンコジーンとして機能することが確認されており、特に膀胱がんにおける主要ドライバー変異として重要です。
尿路上皮がん(膀胱がん)における極めて重要な役割
FGFR3は、ヒトのがんにおいて体細胞変異が最初に報告されたFGFRファミリーメンバーであり、特に膀胱がん(尿路上皮がん:UC)の発症メカニズムにおいて中心的な役割を果たしています。膀胱がんで同定されたFGFR3変異は、主としてエクソン7・10・15に見られ、S249C・Y375C・G380/382R・K650/652Mなど多岐にわたります。特にS249Cは細胞外ドメインの変異であり、膀胱がんにおけるFGFR3変異の約69%を占める最も一般的なものです。
💡 用語解説:NMIBC と MIBC の違い
NMIBC(Non-Muscle-Invasive Bladder Cancer:筋層非浸潤性膀胱がん)は、がんが膀胱の粘膜・粘膜下層にとどまり筋層まで達していない段階です。内視鏡治療(TURBT)が主体で、予後は比較的良好ですが再発しやすいのが特徴です。MIBC(Muscle-Invasive Bladder Cancer:筋層浸潤性膀胱がん)はがんが筋層に達した段階で、根治的膀胱全摘除術や化学療法が必要となり、予後は大幅に悪化します。FGFR3変異はNMIBCに多く、MIBCになると頻度が下がる傾向があります。
大規模な分子プロファイリングによれば、FGFR3変異はNMIBCの39%、局所進行上部尿路上皮がんの43%で確認された一方で、筋層浸潤性がん(MIBC)では14%、転移性がんでは26%と、病期が進行するにつれてその保有頻度が変化します。
尿路上皮がんの進行ステージ別 FGFR3変異保有率
データ出典:PMC (National Center for Biotechnology Information)
膀胱がん
(NMIBC)
上部尿路がん
がん
膀胱がん
(MIBC)
この分布パターンは、膀胱がんには2つの独立した進行経路が存在するという分子病理学的仮説を支持しています。すなわち、染色体9の欠失やFGFR3変異を伴い頻繁に再発を繰り返す「表在性・乳頭状腫瘍経路」と、TP53変異や染色体17のヘテロ接合性喪失(LOH)を伴い急速に進行する「上皮内がん・浸潤性腫瘍経路」です。FGFR3変異は前者の経路において優位です。
治療標的選択において特に注意が必要なのは、原発巣と転移巣の間でFGFR3の変異状態に不一致(Discordance)が生じることです。ある研究では、原発腫瘍と異時性転移を有するFGFR3変異症例27例のうち7例(26%)で変異状態の不一致が認められました。古い原発巣のアーカイブ組織のみに依存して治療標的を選択することの危険性を示す重要な知見です。また、非致死性のFGFR3関連骨格障害(ACHなど)を持つ患者は、すべての細胞にFGFR3変異を有しているため、一般集団よりも膀胱腫瘍などの上皮性腫瘍を発症するリスクが生涯にわたり高い可能性が示唆されています。
多発性骨髄腫(Multiple Myeloma)
💡 用語解説:染色体転座 t(4;14) とは
染色体転座とは、2本の異なる染色体が互いに断片を交換する現象です。多発性骨髄腫に特徴的なt(4;14)(p16.3;q32.3)では、4番染色体のFGFR3遺伝子が14番染色体の強力な免疫グロブリン重鎖(IGH)プロモーターの制御下に置かれます。これによってFGFR3が本来あるべき組織特異的な制御から外れ、骨髄腫細胞で大量かつ恒常的に発現・活性化されることでがん化が促進されます。
血液がんの一種である多発性骨髄腫においても、FGFR3は重要な役割を担います。全症例の約15%において、t(4;14)(p16.3;q32.3)という特定の染色体転座イベントが発生します。この転座により4番染色体上に位置するFGFR3遺伝子が14番染色体上の高度に活性化されている免疫グロブリン重鎖(IGH)プロモーターの制御下に置かれることになります。その結果、FGFR3の過剰発現と恒常的な活性化が促進され、骨髄腫細胞の異常増殖と生存を強力にサポートします。
💡 用語解説:MMSET(NSD2/WHSC1)とは
MMSET(別名 NSD2・WHSC1)は、t(4;14)転座でFGFR3とともに4番染色体上から免疫グロブリン重鎖プロモーターの支配下に移る、もう一つの遺伝子です。ヒストンのメチル化を担うエピジェネティック制御因子で、DNA修復や広範な遺伝子発現を調節します。骨髄腫の悪性化において、FGFR3が「増殖・生存シグナル」を担うのに対し、MMSETは「クローンの増殖・細胞接着の変化・化学療法への耐性」を駆動する、いわば縁の下の主役です。
ここで臨床的に極めて重要なのは、t(4;14)転座で過剰発現する遺伝子はFGFR3だけではないという点です。この転座はFGFR3と同時にMMSET(NSD2/WHSC1)という遺伝子も活性化させます。FGFR3が検出されるのはt(4;14)骨髄腫細胞の約70%にとどまる一方、MMSETはすべてのt(4;14)症例で普遍的に過剰発現しています。つまりFGFR3の発現を失っているサブグループでもMMSETは必ず過剰発現しており、t(4;14)の予後不良を駆動する真の中心はMMSET側にあると考えられています。t(4;14)はボルテゾミブ(プロテアソーム阻害薬)やレナリドミド(免疫調節薬)といった新規薬剤の登場で部分的に改善したものの、依然として強力な独立した予後不良因子であり、これら2剤に二重耐性を示す割合が53%に達するというデータもあります。FGFR3を標的とするだけでは骨髄腫を制御しきれない理由が、ここにあります。
膠芽腫とFGFR3-TACC3融合遺伝子:第3の発がん機序
💡 用語解説:融合遺伝子(Fusion Gene)とは
融合遺伝子(Fusion Gene)とは、本来別々の2つの遺伝子が染色体の切断・再結合によってつなぎ合わさり、ひとつの異常な「ハイブリッド遺伝子」になったものです。そこから作られる融合タンパク質は元の2つのタンパク質の性質を併せ持つため、正常では起こりえない場所ではたらいたり暴走したりします。点変異・染色体転座と並ぶ、第3のがん化のしくみです。
膀胱がんの「点変異」、多発性骨髄腫の「染色体転座」に続く第3のFGFR3異常が、融合遺伝子です。膠芽腫(グリオブラストーマ)をはじめとする一部の脳腫瘍や肺がんでは、4番染色体短腕上で隣り合うFGFR3遺伝子とTACC3遺伝子のあいだに微小な欠失・再構成が起こり、両者がつながったFGFR3-TACC3融合遺伝子が形成されます。この融合遺伝子からは、細胞膜側にFGFR3のキナーゼドメイン、細胞質側にTACC3のコイルドコイルドメインを併せ持つキメラタンパク質が作られます。
通常のFGFR3は細胞膜にとどまって外からのシグナルを受け取る受容体ですが、TACC3由来のコイルドコイルドメインが付加されたこの融合タンパク質は、細胞核内、さらには細胞分裂時の紡錘体極(染色体を引き分ける装置の中心)へと異常に移動する性質を獲得します。本来そこにいないはずのキナーゼが分裂装置に干渉することで、染色体の分配にエラーが生じ、細胞が異数性(染色体の数の異常)を起こしやすくなります。これが強力な発がん能とゲノム不安定性を生み出すしくみです。FGFR3-TACC3融合はFGFR標的薬(チロシンキナーゼ阻害薬)の標的にもなりうるため、膠芽腫などでの検出は治療選択の上でも意味を持ちます。
子宮頸がんとその他の悪性腫瘍
初期の研究報告では子宮頸がんの最大25%でFGFR3変異が見られるとされていましたが、その後の検証研究では51の原発性子宮頸がんと7つの細胞株を対象とした詳細なシークエンス解析の結果、細胞外ドメインのS249C変異が1例のみで確認され、他のすべての腫瘍および細胞株では野生型のFGFR3のみが検出されました。子宮頸がんにおけるFGFR3変異の頻度は当初考えられていたよりもかなり低い可能性が示唆されています。一方で、腎盂・尿管の移行上皮がん、肺がん、精巣がんなど多様な臓器においても体細胞変異が報告されており、多様な上皮系悪性腫瘍の発症メカニズムへの寄与は明らかです。
良性皮膚疾患:脂漏性角化症と表皮母斑
FGFR3変異による細胞増殖作用は悪性腫瘍に限定されるものではありません。表皮母斑(Epidermal Nevi:EN)や脂漏性角化症(Seborrheic Keratoses:SK)といった、悪性化のポテンシャルを持たない良性の増殖性皮膚疾患においても、FGFR3変異が極めて高頻度に同定されています。良性の脂漏性角化症の実に39%でFGFR3の活性化変異が検出されるというデータもあります。
ENでは、FGFR3変異としてR248C(TD I型で知られる変異)が16例中15例で確認されており、同時にPIK3CA変異(E545G)が27%に認められます。SKではより多様な種類のFGFR3変異が見られます。発生時期の観点からも重要な違いがあります。胎児発生の過程で変異が生じた場合は組織のモザイク現象として表皮母斑(EN)を形成し、成人期に体細胞突然変異として生じた場合は局所的な脂漏性角化症(SK)を形成するという、発生時期に依存した明確な表現型の違いが存在します。
5. 診断・遺伝子検査の進め方
骨系統疾患の診断:臨床所見から分子遺伝学的確定まで
ACHの診断は主として臨床的特徴によって行われます。妊娠中の超音波検査では、四肢の短縮(特に前腕・上腕)・大頭・前頭部突出などの所見から出生前に疑われることがあります。出生後は特徴的な骨格所見(根部四肢短縮・大頭・顔面中央部低形成など)に加え、骨のX線所見(腸骨の変形・基底部狭小・長管骨の短縮など)を組み合わせて臨床診断します。分子遺伝学的確定はFGFR3遺伝子の標的変異解析(c.1138G>A/C)によって行われ、ほぼ100%の感度を持ちます。より重症のTDやSADDAN、あるいは非典型的症例では、FGFRパネル検査や全エクソーム解析(WES)が用いられます。
出生前診断の選択肢として、すでにFGFR3変異を有するACH患者やキャリアからの次子については、絨毛検査・羊水検査による出生前遺伝子診断が可能です。確定診断後の適切な分娩計画(帝王切開)の立案にも遺伝情報は不可欠です。
がん治療におけるFGFR3遺伝子検査の位置づけ
膀胱がんをはじめとするFGFR3変異陽性腫瘍の同定には、腫瘍組織の次世代シーケンス(NGS)パネル検査が使用されます。エルダフィチニブなどのFGFR標的治療薬を使用する際には、FGFR2またはFGFR3の遺伝子改変が必須の適応条件となっています。治療中の耐性獲得モニタリングには、血漿中の細胞遊離DNA(cfDNA)を用いたリキッドバイオプシーが活用されます。
💡 用語解説:リキッドバイオプシー(cfDNA検査)とは
リキッドバイオプシーとは、血液などの体液から採取できる細胞遊離DNA(cfDNA)や循環腫瘍DNA(ctDNA)を解析することで、腫瘍の遺伝情報をリアルタイムで把握する手法です。従来の組織生検と異なり低侵襲で繰り返し実施でき、治療中に獲得された耐性変異(ゲートキーパー変異など)を早期に検出できます。FGFR3陽性膀胱がんの治療モニタリングにおいて、この技術は耐性メカニズムの早期把握と次治療への迅速な移行に貢献しています。
前述のとおり、原発巣と転移巣の間でFGFR3変異状態に26%の不一致が生じることが報告されています。このことから、最新の転移組織またはcfDNAを用いた再生検・再評価が、治療開始前および増悪時に重要となります。古い腫瘍組織のみに頼った治療標的選択には限界があることを臨床医は認識する必要があります。
6. 分子標的治療の最前線
FGFR3の過剰活性化を特異的かつ安全に抑制することは、軟骨無形成症の根治的治療およびFGFR3駆動性がんの治療において最も重要かつ挑戦的な課題です。近年、細胞内シグナルの下流経路を阻害するペプチドアナログ製剤や、受容体自体を選択的に阻害する次世代のチロシンキナーゼ阻害薬(TKI)の開発が急速に進展しており、臨床試験において画期的な成果を上げています。
CNPアナログ:骨系統疾患への初の薬物療法
💡 用語解説:CNP/NPR2/cGMP経路とは
CNP(C型ナトリウム利尿ペプチド)は、グアニル酸シクラーゼ受容体であるNPR2(GC-B)に結合してcGMPを産生するシグナル分子です。cGMPはプロテインキナーゼG II(cGKII)を活性化し、これがFGFR3の下流にあるRAF1の活性化を機能的に拮抗(ブロック)します。つまり、FGFR3を直接止めるのではなく、「ブレーキに対抗する別のブレーキ解除システム」を増強することで骨成長を促進する間接的なアプローチです。CNPアナログはこのメカニズムを活用した初の実用化された骨成長促進薬です。
ボソリチド(Vosoritide / 製品名:Voxzogo)は、BMN111としても知られるCNPアナログ(1日1回皮下注射)です。生体内のCNPにアミノ酸を追加して中性エンドペプチダーゼに対する耐性を持たせることで血漿半減期を延長しています。米国・欧州・ブラジルでは2021年、日本では2022年6月にACHの初の特異的薬物療法として承認され、現在では生後から成長板が閉鎖するまでの小児患者において広範に使用可能となっています。日本では「骨端線閉鎖を伴わない全年齢の小児の軟骨無形成症」を適応とする希少疾病用医薬品として承認されており、年齢の下限が設けられていない点が特徴で、約1,500人の患者が対象と見込まれています。なお、ボソリチドの基礎となったCNP(C型ナトリウム利尿ペプチド)は、1990年に日本で骨成長の天然の調節因子として初めて発見されたペプチドです。15 µg/kgの投与により第II相試験では2歳以上の小児において年間成長速度(AHV)および身長の顕著な改善(未治療群と比較して+6.3〜+7.8cmの絶対的な身長獲得)が確認されました。長期的なデータでも、3年間の継続治療により脊柱の矢状面バランスの改善・脚の湾曲(O脚)の減少・身体的な健康関連QOLの向上が報告されています。全世界での売上は承認からわずか1年後の2022年時点で約1億6,900万ドルに達しており、2029年には13億ドルに達すると予測されています。
ナベペグリチド(Navepegritide / TransCon CNP)は、患者家族の日々の注射負担をさらに軽減するために開発されたCNPプロドラッグです。CNP-38が切断可能なリンカーを介してPEG(ポリエチレングリコール)キャリア分子に結合しており、生体内で徐放されることで週1回の投与で十分な有効血中濃度を維持できます。米国では2026年2月にYUVIWEL®(ユビウェル)の製品名で2歳から成長板閉鎖までの小児を対象に承認されました(日本は2026年5月時点で未承認)。2031年までに5億9,200万ドルの売上が見込まれています。
経口TKI:注射からの解放と骨格プロポーション改善
💡 用語解説:チロシンキナーゼ阻害薬(TKI)とは
チロシンキナーゼ阻害薬(TKI:Tyrosine Kinase Inhibitor)とは、受容体チロシンキナーゼの細胞内ドメインに入り込み、ATP結合部位をブロックすることで受容体のリン酸化(活性化)を直接阻害する低分子化合物です。CNPアナログが下流シグナルで間接的に拮抗するのとは異なり、TKIは受容体自体のシグナル発生源を根本からブロックします。何より経口投与が可能であることは、毎日の注射を強いられる患者・家族の精神的・物理的負担を根本から解消する画期的な利点です。
インフィグラチニブ(Infigratinib)は、FGFR1〜3を標的とする経口低分子阻害薬です。BridgeBio社が実施した第III相無作為化臨床試験(PROPEL3試験:NCT06164951)において、プラセボ群と比較してベースラインからの年間成長速度(AHV)の統計的に有意かつ臨床的に意義のある増加(+2.10 cm/年)を示し、主要評価項目を達成しました。52週時点での絶対成長量は、プラセボ群が4.22 cm/年であったのに対し、インフィグラチニブ群は5.96 cm/年でした。
インフィグラチニブによる年間成長速度の改善(PROPEL3試験)
データ出典:Clinical Trials Arena
第III相無作為化臨床試験(PROPEL3)において、インフィグラチニブ投与群の年間成長速度が統計的に有意に改善(+2.10 cm/年)。
さらに特筆すべき点として、8歳未満の小児を対象とした事前定義の探索的解析では、上半身と下半身のプロポーション(体型比)の統計的に有意な改善(LS平均減少量0.05)も確認され、無作為化試験でこの「プロポーションの改善」マイルストーンに到達した史上初の薬剤となりました(ただし全体集団では有意差には達していない)。本剤は忍容性が高く、重篤な副作用や治療関連の投与中止は報告されていません。高リン血症も軽度なものが3例のみで減量には至らず、眼科系副作用も観察されていません。2026年後半には規制当局へ新薬承認申請(NDA/MAA)が提出される予定です。
ダボグラチニブ(Dabogratinib / TYRA-300)は、Tyra Biosciences社が開発中のFGFR3に対して極めて高度な「選択性」を有する次世代の経口阻害薬です。従来のTKIが抱えていた最大の課題(FGFR1:リン酸代謝への関与、FGFR2・FGFR4の同時阻害による毒性)を回避するよう精密に設計されており、2024年10月に米国FDAから治験新薬(IND)申請のクリアランスを受けました。小児のACH患者(3〜10歳)を対象とした第2相臨床試験(BEACH301試験)が2025年第1四半期に開始される予定です。治療未経験者(コホート1)および治療歴のある者(コホート2)を対象とし、0.125〜0.50 mg/kgの複数の用量レベルで最大12ヶ月の投与を行います。安全性・年間成長速度の改善に加え、機能的改善(リーチ・歩行)や脊椎疾患の改善といったQOLに直結する探索的評価項目も設定されています。
開発中止に終わったアプローチ:レシフェルセプト(デコイ受容体)
💡 用語解説:デコイ受容体(おとり受容体)とは
デコイ受容体(Decoy Receptor/おとり受容体)とは、受容体のうちリガンドと結合する部分だけを取り出した「おとり」を体内に投与し、過剰なシグナル分子(リガンド)を先回りして捕まえてしまう治療戦略です。レシフェルセプトはFGFR3の細胞外ドメインをおとりとして使い、余分なFGFリガンドを吸着することで、本物のFGFR3が過剰に活性化されるのを防ごうとしました。
レシフェルセプト(Recifercept / PF-07256472)は、ファイザー社が開発していた注射用の組換えタンパク質製剤です。FGFR3の細胞外ドメインを「おとり受容体」として用い、過剰なFGFリガンドを捕捉して内因性FGFR3への結合を防ぐという、ボソリチド(CNP経路)やTKI(受容体阻害)とはまったく異なる第4のアプローチでした。しかし、生後15ヶ月〜12歳未満のACH患児を対象とした第2相試験(C4181005試験)で、約45人が6ヶ月間の治療を受けた時点の中間解析の結果、プラセボ(歴史的対照群)と比較して事前に設定された有効性基準(身長の増加)を満たせませんでした。安全性に大きな懸念はなかったものの、この有効性不足を受けて、ファイザー社は2022年11月に本プログラムのグローバル開発の早期終了を決定し、長期継続試験も中止されました。
この結果は、FGFR3関連疾患の治療がすべて順調に進んでいるわけではないことを示す重要な教訓です。FGF受容体とリガンドのネットワークへの介入は、生体内の代償機構によって効果が相殺されるリスクを常にはらんでおり、「どの分子を・どう狙うか」という標的戦略の選定が成否を分けます。同じACHを標的にしながら、ボソリチドやインフィグラチニブが成功しレシフェルセプトが届かなかったという対比は、精密医療における標的選定の難しさを物語っています。
既存薬のリポジショニングとその他のアプローチ
新薬開発と並行して、既存薬のFGFR3経路への影響も研究されています。一般的な抗ヒスタミン剤(制吐剤)であるメクロジン(Meclozine)は、ERK1/2のリン酸化をダウンレギュレートすることでFGFR3の下流シグナルを抑制し、軟骨細胞の増殖と分化を促進することがスクリーニングによって発見されました。ACHのマウスモデルで骨の長軸成長を促進し、小児患者への投与試験でも重篤な副作用を引き起こさないことが確認されており、安価で安全な代替療法としての可能性を秘めています。他にも、カカオ由来のフェノール化合物である(-)-エピカテキンによる一次繊毛の異常是正効果、スタチンによるFGFR3分解促進作用、PTH(1-34)治療によるFgfr3発現抑制と軟骨細胞増殖増加作用などが基礎研究レベルで有望な候補として検討されています。
がん治療におけるFGFR標的治療:現状と課題
💡 用語解説:ゲートキーパー変異とは
ゲートキーパー変異(Gatekeeper mutation)とは、標的阻害薬への曝露によって選択された二次的な耐性変異で、阻害薬がキナーゼのATP結合部位に物理的に結合できなくなる変異です。FGFR3の場合、エルダフィチニブ治療中にV555M変異などが出現します。これは第一世代TKIの主要な獲得耐性メカニズムのひとつであり、次世代阻害薬(TYRA-300など)はこの変異に対しても効力を維持するよう設計されています。
エルダフィチニブ(Erdafitinib)は、FGFR2またはFGFR3の遺伝子改変を有する転移性尿路上皮がん(mUC)に対して承認されている唯一のFGFR標的療法です。米国(2024年1月)・欧州(2024年8月)に続き、日本でも2024年12月27日に「がん化学療法後に増悪したFGFR3遺伝子変異又は融合遺伝子を有する根治切除不能な尿路上皮癌」を適応として承認され、2025年7月に薬価収載・発売されました。これは尿路上皮がんに対する日本初・唯一の、遺伝子異常に基づく治療薬(FGFR阻害薬)です。一方、前向きに収集された臨床データによれば、客観的奏効率(ORR)は40%を示す一方で、無増悪生存期間(PFS)の中央値はわずか2.8ヶ月、全生存期間(OS)の中央値は6.6ヶ月にとどまるという重大な限界が露呈しています。毒性の面では患者の83.7%で高リン血症が発現し(FGFR1の同時阻害による)、口内炎(69.8%)・口腔乾燥(37.2%)などの有害事象も頻発します。患者の50%が投与中断を、38%が用量減量を余儀なくされており、十分な用量強度を維持することが困難です。また、治療中にTP53・AKT1などのバイパス経路の変異やゲートキーパー変異(V555Mなど)が獲得され、cfDNAリキッドバイオプシーでリアルタイムに検出されることが確認されています。
この状況を打破するゲームチェンジャーとして期待されているのがダボグラチニブ(TYRA-300)です。FGFR3への極めて高い選択性を持つため、エルダフィチニブで問題となったFGFR1/2阻害に関連する高リン血症や毒性を大幅に軽減できます。さらに重要なことに、ゲートキーパー変異(V555M/L)の影響を受けずにFGFR3に結合し効力を維持できるという分子構造上の画期的な強みを持っています。進行性固形がん・mUCを対象とした第1/2相試験(SURF301試験:NCT05544552)の中間データでは、重度に前治療を受けたFGFR3陽性mUC患者のうち90mg/日以上の用量で投与された11例中6例(54.5%)が確認済みの部分奏効(PR)を達成し、病勢コントロール率(DCR)は100%に達しました。すべての用量においてFGFR1/FGFR2関連の毒性の発現は非常に稀であり、安全性プロファイルも良好でした。
| 薬剤名 | クラス・作用機序 | 投与経路 | 開発状況 | 主な特徴・利点 |
|---|---|---|---|---|
| ボソリチド | CNPアナログ(NPR2を介した拮抗) | 皮下注射(1日1回) | 承認済み | 初の承認薬、3年長期データでQOL改善を確認 |
| ナベペグリチド | CNPプロドラッグ | 皮下注射(週1回) | 承認済み | 週1回投与で患者・家族の負担を軽減 |
| インフィグラチニブ | 汎FGFR阻害薬(経口TKI) | 経口(1日1回) | 第III相完了・申請準備中 | 注射不要、プロポーション改善(史上初) |
| ダボグラチニブ(TYRA-300) | FGFR3選択的TKI(次世代) | 経口 | 第II相開始(BEACH301) | FGFR1/2毒性回避、ゲートキーパー変異克服 |
| エルダフィチニブ | 汎FGFR阻害薬(がん治療用) | 経口 | 承認済み(mUC) | 唯一FDA承認のmUC標的療法、ORR 40% |
| メクロジン | 抗ヒスタミン剤(ERK抑制) | 経口 | 臨床研究段階 | 既存薬リポジショニング、安全性プロファイル良好 |
| レシフェルセプト | デコイ受容体(FGFリガンド捕捉) | 皮下注射 | 開発中止 | 第2相で有効性基準未達、2022年11月に開発終了 |
7. 遺伝カウンセリングの意義
FGFR3関連疾患の確定診断後、患者・家族への丁寧な遺伝カウンセリングが不可欠です。疾患の希少性・重症度の多様性・再発リスク・治療選択肢の急速な進化という文脈の中で、個々の家族が十分な情報に基づいた意思決定ができるよう支援することが遺伝カウンセリングの本質的な目標です。
💡 用語解説:常染色体優性遺伝(顕性遺伝)とは
常染色体優性遺伝(顕性遺伝)とは、性染色体以外の常染色体上に存在する変異アレルが1コピーだけで疾患を引き起こす遺伝形式です。患者が子どもを持つ場合の遺伝確率は理論上50%です。FGFR3関連骨系統疾患の多くはこの遺伝形式をとりますが、特にACHでは約80%がde novo(新規)変異であるため、罹患者の両親が健康であっても子どもに疾患が発生します。これは「親から遺伝した」ことを意味せず、突然変異によるものです。父親の加齢(高齢父親)がde novo変異リスクを高めることが知られています。
- ➤遺伝形式と再発リスクの説明:ACHの多くはde novo変異であり、親への遺伝は認められません。患者本人が子どもを持つ場合の遺伝確率は理論上50%です。ACH同士の妊娠では25%の確率でホモ接合体(致死的)が生まれるリスクがあるため、特に慎重な遺伝カウンセリングが必要です。
- ➤出生前診断の選択肢:FGFR3変異が既知の場合は絨毛検査・羊水検査による出生前遺伝子診断が可能です。確定診断後は早期から分娩方法(帝王切開)の計画を立て、新生児期の大後頭孔狭窄モニタリング体制を整えることが重要です。
- ➤長期リスクモニタリング:ACH患者はすべての細胞にFGFR3変異を有しているため、膀胱がんなどの上皮性腫瘍発症リスクが一般集団より高い可能性があります。定期的な泌尿器科的フォローアップを考慮することが望ましいとされています。
- ➤治療と意思決定のサポート:ボソリチドなど新薬が承認されている現在、患者が十分な情報に基づいて「いつ・どの治療を選ぶか」を自律的に決定できるよう支援することが遺伝カウンセリングの中核となっています。不可逆的な骨延長術は、患者自身が意思決定できる年齢まで延期することが多くの専門家によって推奨されています。
8. よくある誤解
誤解①「軟骨無形成症は知的障害を伴う」
これは誤りです。ACH患者の知的発達は通常正常範囲にあり、学習能力・認知機能に影響を与えません。運動発達マイルストーンの遅れが見られることはありますが、それは骨格的な制約によるもので、知的発達とは切り離して考える必要があります。
誤解②「FGFR3変異があれば必ずがんになる」
誤りです。骨系統疾患を引き起こす生殖細胞系列変異と、がんを引き起こす体細胞変異は発生する細胞・組織が異なります。ACH患者は一般集団よりリスクが高い可能性が示唆されてはいますが、自動的にがんを発症するわけではありません。
誤解③「ACHの治療は手術(骨延長術)しかない」
現在は誤りです。ボソリチド(2021年承認)やナベペグリチドといった薬物療法が第一選択として確立されつつあります。多くの専門家は、患者が自律的に意思決定できる年齢まで不可逆的な外科的介入を延期することを推奨しています。
誤解④「ACHは必ず親から遺伝する」
これは誤りです。ACH患者の約80%はde novo(新規)変異によるものであり、両親はいずれも変異を持ちません。「両親が健康なのになぜ?」という疑問は非常によくありますが、自然発生した新規変異が原因であることがほとんどです。誰の「せい」でもありません。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 FGFR3関連疾患・遺伝カウンセリングについて
軟骨無形成症をはじめとするFGFR3関連疾患のご相談、
出生前診断・遺伝子検査に関するお問い合わせは、
臨床遺伝専門医が在籍するミネルバクリニックへ。
関連記事
参考文献
- [1] MedlinePlus Genetics. FGFR3 gene. National Library of Medicine. [MedlinePlus]
- [2] Pauli RM. Achondroplasia. In: Adam MP, et al., editors. GeneReviews®. Seattle: University of Washington; 1998 [updated 2023]. [NCBI Bookshelf]
- [3] Ornitz DM, Marie PJ. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 2015;29(14):1463-1486. [PMC4526732]
- [4] Foldynova-Trantirkova S, et al. FGFR3 signal transduction in chondrocytes and mechanisms of FGFR3 inhibitors. PLOS ONE. 2012. [PLOS figshare]
- [5] Chesi M, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma. Reactome FGFR3 mutant receptor activation. [Reactome]
- [6] Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer. 2017;17(5):318-332. [PMC3809064]
- [7] Cappellen D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23(1):18-20. [PubMed]
- [8] Tomlinson DC, et al. FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. J Pathol. 2007. [PMC1891972]
- [9] Kamat AM, et al. Clinical and genomic landscape of FGFR3-altered urothelial carcinoma and treatment outcomes with erdafitinib: a real-world experience. NPJ Precis Oncol. 2024. [PMC11233068]
- [10] Hafner A, et al. FGFR3 and Tp53 mutations in T1G3 transitional bladder carcinomas: independent distribution and lack of association with prognosis. Clin Cancer Res. 2005;11(15):5444-5450. [AACR Journals]
- [11] Hafner C, et al. Mosaicism of activating FGFR3 mutations in human skin causes epidermal nevi. J Clin Invest. 2006;116(8):2201-2207. [JCI]
- [12] Xie Y, et al. Achondroplasia: Development, Pathogenesis, and Therapy. Front Genet. 2017;7:219. [PMC5354942]
- [13] BridgeBio Pharma. Phase III PROPEL3 trial results for infigratinib in achondroplasia. Clinical Trials Arena. 2025. [Clinical Trials Arena]
- [14] Tyra Biosciences. Tyra Biosciences receives IND clearance from FDA to proceed with Phase 2 BEACH301 trial of TYRA-300 in achondroplasia. 2024. [Tyra Biosciences IR]
- [15] Tyra Biosciences. Interim clinical proof-of-concept data for TYRA-300 in FGFR3-positive metastatic urothelial carcinoma. 2024. [Tyra Biosciences PDF]
- [16] OMIM #100800. Achondroplasia. Johns Hopkins University. [OMIM]






