目次

ASXL3遺伝子は、18番染色体長腕(18q12.1)に位置し、脳の発達やシナプス機能に必須のエピジェネティック制御因子をコードしています。BAP1という酵素と複合体を形成し、ヒストン修飾を介して遺伝子のオン・オフを精密に調整する「司令塔」のような働きを担っています。この遺伝子に機能喪失型変異が生じると、Bainbridge-Ropers症候群(BRPS)と呼ばれる重度の神経発達障害を引き起こすほか、非症候群性の自閉症スペクトラム障害(ASD)の強力なリスク遺伝子としても注目されています。
Q. ASXL3遺伝子とはどのような遺伝子ですか?まず結論だけ知りたいです
A. 18番染色体(18q12.1)上にあり、BAP1という酵素と組んで「PR-DUB複合体」を形成し、ヒストンの修飾を介して脳の発達に必須な遺伝子のスイッチを精密にコントロールするエピジェネティック制御因子です。機能喪失型変異が生じると、重度の発達遅滞・特徴的な顔貌・摂食障害を伴うBainbridge-Ropers症候群を引き起こすほか、自閉症スペクトラム障害の強力なリスク遺伝子としても認識されています。
- ➤基本情報 → HGNC:29357、NCBI:80816、OMIM:615115、染色体座位18q12.1、2,248アミノ酸のスキャフォールドタンパク質
- ➤分子機能 → BAP1と結合してPR-DUB複合体を形成、ヒストンH2AK119のユビキチンを除去
- ➤変異の特徴 → 病原性変異の99%がエクソン11・12に集中、ハプロ不全が主な発症機序
- ➤関連疾患 → Bainbridge-Ropers症候群(OMIM #615485)、非症候群性ASDの中核リスク遺伝子
- ➤検査の選択肢 → トリオ全エクソーム解析・知的障害遺伝子パネル・NIPTインペリアルプラン
1. ASXL3遺伝子の基本情報
ASXL3(Additional sex combs-like 3)遺伝子は、ヒトの18番染色体長腕12.1領域に位置する、進化的に高度に保存された転写調節因子をコードする遺伝子です。ショウジョウバエの「Additional sex combs(Asx)」遺伝子のヒト相同遺伝子ファミリー(ASXL1・ASXL2・ASXL3)の一員であり、クロマチンリモデリングとエピジェネティックな遺伝子発現制御の中心ハブとして、特に脳の発達において欠かせない役割を担っています[1]。
💡 用語解説:エピジェネティクスとは
エピジェネティクスとは、DNAの塩基配列そのものは変えずに、遺伝子の「読まれ方(オン・オフ)」を制御する仕組みのことです。DNAそのものが「楽譜」だとすると、エピジェネティクスは「どのページを開いて、どこを演奏するか」を決める指示書のようなものです。代表的な仕組みにはDNAのメチル化やヒストン(DNAを巻きつけるタンパク質)の化学修飾があり、ASXL3はこのヒストン修飾を介して遺伝子のスイッチを精密に制御しています。
臨床医学の文脈では、ASXL3遺伝子の機能喪失型変異は2013年にBainbridgeらによって初めて報告され、重度の知的障害・広範な発達遅滞・顕著な筋緊張低下・特徴的な顔貌・重篤な摂食障害を中核症状とするBainbridge-Ropers症候群の原因として特定されました[2]。その後の大規模ゲノム解析により、ASXL3は英国DDD研究(9,625例の知的障害トリオ解析)で最も頻繁に変異が見つかる上位10遺伝子に含まれる(約1:193の頻度)こと、さらに非症候群性の自閉症スペクトラム障害(ASD)の中核リスク遺伝子としても機能することが解明され、神経発達障害領域における極めて重要な遺伝子と位置づけられています[3]。
2. ASXL3タンパク質の分子構造と多面的機能
ASXL3がコードするタンパク質は、約2,248個のアミノ酸からなる巨大な「足場(スキャフォールド)タンパク質」です[4]。ASXL3自体は酵素活性を持っていません。その代わりに、複数の高度に保存されたドメインを介して他の修飾酵素や転写因子と複合体を形成し、ゲノム上の特定の領域にエピジェネティックな変化を誘導する「ハブ」として機能します。この多機能性こそが、ASXL3が細胞分化や組織発生において広範な影響を及ぼす理由です。
主要な4つの構造ドメインとその役割
🧭 ASXNドメイン(HARE-HTH)
タンパク質のN末端側に位置し、DNAへの直接結合を促進。複合体を標的のゲノム領域へ正確に運ぶ「カーナビ」のような役割を担うと予測されています。
🔗 ASXHドメイン(DEUBAD)
エクソン9〜11にまたがる機能の核心領域。脱ユビキチン化酵素であるBAP1(BRCA1 Associated Protein 1)と強固に相互作用し、PR-DUB複合体の中核を形成します。
⚙️ ASXM1・ASXM2ドメイン
核内受容体との結合を媒介。オキシステロール受容体LXR-αや甲状腺ホルモン受容体TR-βに結合して脂質新生(lipogenesis)を負に制御する機能が確認されています。
📖 PHDフィンガードメイン
C末端のジンクフィンガードメイン。クロマチン上の特定のヒストン修飾を直接認識する「エピジェネティック・リーダー」として機能します。
💡 用語解説:スキャフォールドタンパク質とは
「スキャフォールド(scaffold)」とは「足場」という意味で、自分自身は酵素活性を持たないものの、複数のタンパク質を物理的に集めて結びつける「土台」となるタンパク質のことを指します。建築現場の足場が職人を支えるのと同じように、ASXL3はBAP1などの活性酵素を正しい場所・正しいタイミングで働かせるための舞台装置として機能します。この「土台」が壊れると、酵素自体が無事でも複合体全体の機能が成り立たなくなります。
さらにASXL3は、転写伸長を促進するBRD4タンパク質とBAP1を物理的につなぐ「アダプター」としても機能し、活性化されたエンハンサー領域での転写活性化状態を持続させる重要な役割を担います[4]。
3. PR-DUB複合体:エピジェネティック制御の核心メカニズム
神経発生過程においてASXL3が果たす最も重要な働きは、BAP1と結合して形成する「ポリコーム抑制性脱ユビキチン化(PR-DUB)複合体」の機能にあります[5]。この複合体の働きを理解することが、関連疾患の病態理解への近道です。
💡 用語解説:ヒストンとユビキチン化
ヒストンとは、長いDNAをコンパクトに折りたたむために巻きつけるタンパク質の「糸巻き」のような存在です。ユビキチン化とは、このヒストンタンパク質に「ユビキチン」という小さなタグを付ける化学修飾で、遺伝子の働きをオン・オフする「目印」として機能します。ヒストンH2Aのリジン119にユビキチンが1つ付いた状態(H2AK119ub1)は「遺伝子オフ」の合図であり、これが過剰に溜まるとクロマチンが固く凝集して、本来発現すべき遺伝子が抑え込まれてしまいます。
細胞内では、ポリコーム抑制複合体1(PRC1)がヒストンH2Aのリジン119にユビキチンを付ける(H2AK119ub1)ことでクロマチンを凝集させ、遺伝子発現を抑制します。これに対し、PR-DUB複合体の触媒サブユニットであるBAP1は、ASXL3という足場に依存してこのユビキチンを特異的に除去(脱ユビキチン化)します[5]。このユビキチンの「付与」と「除去」のバランスこそが、遺伝子のスイッチを精密に制御する根本メカニズムなのです。
ASXL3が壊れたとき何が起きるか
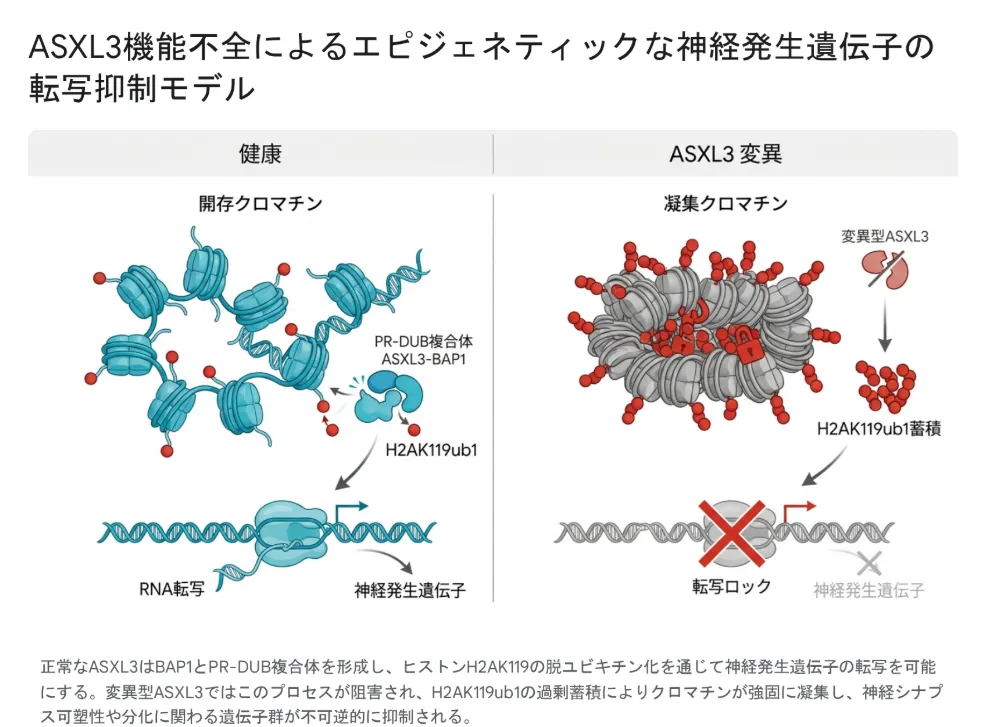
正常なASXL3はBAP1とPR-DUB複合体を形成し、ヒストンH2AK119の脱ユビキチン化を通じて神経発生遺伝子の転写を可能にします。変異型ASXL3ではこのプロセスが阻害され、H2AK119ub1の過剰蓄積によりクロマチンが強固に凝集し、神経シナプス可塑性や分化に関わる遺伝子群が抑制されます。
実際にBainbridge-Ropers症候群の患者さん由来の線維芽細胞を用いた研究では、ゲノム全体のH2AK119ub1レベルが健常な対照細胞の約5倍にまで異常上昇していることが確認されました[5]。さらにトランスクリプトーム解析では564個もの遺伝子に発現変動(うち52%がアップレギュレート、48%がダウンレギュレート)が見られ、転写制御・発生・細胞増殖に関わる遺伝子群が広範に巻き込まれていました。この大規模なエピジェネティックの破綻こそが、脳の発達を根底から狂わせる中心的な病態生理学的機序です。
4. ASXL3変異の特徴と病的メカニズム
ASXL3遺伝子の病原性変異には、いくつかの非常に特徴的なパターンがあります。これらを正確に理解することは、診断の精度と遺伝カウンセリングの質を大きく左右します。
変異の99%は「2か所」に集中する
ASXL3の病原性変異のうち、実に99%がエクソン11とエクソン12内の2つの「変異クラスター領域(Mutation Cluster Regions: MCRs)」に集中していることが明らかになっています[6]。さらに、ナンセンス変異やフレームシフト変異といったタンパク質の途中で切れてしまう「トランケーティング変異」が大部分を占め、ミスセンス変異は比較的少数派です。
💡 用語解説:ハプロ不全(ハプロインサフィシエンシー)とは
人は通常、父と母から1本ずつ計2本の遺伝子を受け継いでいます。「ハプロ不全」とは、2本のうち1本に変異が生じてタンパク質が作れなくなるだけで、必要な量の半分しか確保できず、症状が出てしまう状態を指します。ASXL3の場合、片方のアレル(遺伝子のコピー)が機能を失うだけで、正常な発育に必要な閾値量を満たせず、疾患を発症します。この遺伝子は機能喪失型変異に対して極めて非寛容(intolerant)であることが、大規模ゲノムデータベース(gnomAD等)の指標からも示されています。
エクソン11とエクソン12で違いがある(遺伝子型・表現型相関)
💡 用語解説:NMD(ナンセンス変異依存ミスマッチ修復機構)
細胞には、異常な「設計図(mRNA)」を見つけ次第ただちに分解する品質管理システムが備わっています。これがNMD(Nonsense-Mediated mRNA Decay)です。途中で終わってしまう異常なmRNAは、変な短いタンパク質を作って害を及ぼす前にNMDによって除去されます。ただし、最終エクソンの末端付近で生じた変異はNMDの目を逃れることがあり、結果として不完全なタンパク質が細胞内に残ってしまうことがあります。
5′ MCR(エクソン11の変異)
トランケーティング変異により異常mRNAが作られ、NMDによって速やかに分解。変異タンパク質はほとんど作られません。
臨床的特徴:低身長、重度の小頭症、重度の発達遅延、アーチ状の眉などの顔貌異常がより目立つ傾向。
3′ MCR(エクソン12の変異)
遺伝子末端付近のため、NMDの分解を免れることが多く、C末端のPHDフィンガーなどを欠いた短縮型タンパク質が細胞内に残ります。
臨床的特徴:周産期の重篤な摂食障害、消化器症状、先天性心疾患(心室中隔欠損など)を伴う症例の多くがこの領域から報告されています。
de novo変異とモザイク現象
Bainbridge-Ropers症候群は常染色体顕性(優性)遺伝疾患として分類されますが、報告されているほぼすべての症例はde novo(新生)変異に起因します。つまり両親には変異が存在せず、患者さん本人で初めて生じた変異です[7]。
近年、臨床的に無症状の親の末梢血DNAから低レベルのモザイク変異(バリアントアレル頻度約15%)が検出され、それが複数の子どもに遺伝してBainbridge-Ropers症候群を発症させた家族例も報告されています[8]。これは遺伝カウンセリングにおいて親のモザイク現象の評価が極めて重要であることを示しています。「両親が健康だから次の子も大丈夫」と単純に判断することはできません。
5. ASXL3関連疾患:Bainbridge-Ropers症候群と非症候群性ASD
ASXL3変異に起因する代表的な疾患がBainbridge-Ropers症候群(BRPS、OMIM #615485)です。中枢神経系のみならず、骨格・消化器・循環器など多系統にわたる症状を呈する、非常に異質性の高い症候群です[2]。
Bainbridge-Ropers症候群の主な特徴
🧠 神経・発達
- 全面的発達遅滞・知的障害:97%
- 重度の言語遅延または無発語
- 歩行開始の遅延(19〜20か月以降)
😋 摂食・消化器
- 深刻な摂食困難:78%
- 重度の胃食道逆流症(GERD)
- 周期性嘔吐・誤嚥性肺炎リスク
👤 顔貌・身体
- 特徴的顔貌:98%
- 突出した前額部・アーチ状の眉
- 反り返った下唇・管状の鼻
💪 筋骨格・運動
- 乳児期の筋緊張低下:88%
- 痙縮への移行・関節拘縮
- 側弯症・マルファン様体型

非症候群性ASDの強力なリスク遺伝子
ASXL3が引き起こすのはBainbridge-Ropers症候群のような重篤な多系統症候群だけではありません。広範な身体的奇形を伴わない「非症候群性ASD」の強力なリスク遺伝子としても機能します[3]。
SPARKコホートなどを含む35,000件以上のASD症例を用いた大規模なメタ解析(TADA等)において、ASXL3は全エクソームレベルの有意水準(P < 2.5E-06)およびFDR ≤ 0.01(95%の確率で真の自閉症遺伝子)に到達しており、シナプス形成や転写制御に関わる中核的なASD遺伝子ネットワークの一部を構成していることが実証されています。Bainbridge-Ropers症候群の患者さんでも約30〜78%がASDの正式な診断基準を満たすか、強い自閉症特性(常同行動・視線の合いにくさ・自傷行為など)を示します。
6. モデル生物が明かした発生プロセスへの影響
ASXL3がなぜこれほど広範かつ重篤な神経・身体的欠陥を引き起こすのかを細胞・組織レベルで解明するため、強力な動物モデルを用いた発生生物学的研究が進められています。
アフリカツメガエル胚モデル
研究者らがアフリカツメガエル初期胚でASXL3タンパク質をノックダウンしたところ、細胞がどの組織になるかを決める「神経細胞の運命決定」プロセスが根底から崩壊することが判明しました[9]。前脳・後脳・初代ニューロン、そして顔面や末梢神経系の基となる神経堤の正常な形成に必要な遺伝子群の発現が著しく低下し、Bainbridge-Ropers症候群の患者さんで見られる頭蓋顎顔面の異常や後脳の発達障害と驚くほど一致した表現型が再現されました。これはASXL3が脊椎動物の初期神経系形成における「マスターレギュレーター」として働くことを証明しています。
マウスモデル(Asxl3fs/fs)が示す大脳皮質形成の異常
トランケーティング変異を模倣したマウスモデルでは、大脳皮質の形成において以下のような細胞レベルでの異常が観察されました[10]。
- ➤側方拡張の異常と神経前駆細胞の過剰:本来は分化すべきタイミングで分化できず、前駆細胞が増えすぎた状態が続きます。
- ➤深層ニューロンの分化遅延:大脳皮質6層構造のうち深層を形成するニューロンの分化が著しく遅延・阻害されます。
- ➤シグナル伝達と細胞組成の変容:神経細胞の配置を誘導するシグナル経路が崩壊し、最終的な脳のネットワーク構造が変わってしまいます。
これらの所見は、ASXL3の機能喪失が単なる細胞増殖の停止ではなく、脳の配線計画や細胞アイデンティティ決定の「タイミング」を根底から狂わせる発生プログラムの進行エラーであることを明確に示しています。
7. ASXL3遺伝子検査:いつ・どの検査を選ぶか
ASXL3関連障害には特異的な生化学マーカーは存在せず、顔貌の異常も非特異的であるため、臨床的推測のみで確定診断することは困難です。分子遺伝学的検査が確定診断の唯一の手段となります[2]。
ミネルバクリニックでASXL3を解析できる検査プラン
💡 用語解説:トリオ全エクソーム解析(Trio-WES)
WES(Whole Exome Sequencing)とは、遺伝子のタンパク質をコードする領域(エクソン)全体を網羅的に解析する次世代シーケンス手法です。「トリオ」とは患者さん本人だけでなく両親も含めた3名で同時解析することを指します。Bainbridge-Ropers症候群のようにde novo変異が多い疾患では、両親と比較することで「子どもにだけ新しく生じた変異」を効率的に検出できるため、最も精度の高い診断アプローチとなります。
なお、ミネルバクリニックでは臨床遺伝専門医が検査の選択から結果解釈、その後の遺伝カウンセリングまで一貫してサポートします。RNA統合シークエンス解析(RNA-ISE)を除き、ほとんどの検査はオンライン診療で完結でき、全国の提携施設での採血が可能です。
8. 最新の研究動向と将来の治療展望
現在のところASXL3変異そのものを修正する根本的治療法は存在せず、症状に応じた個別管理(対症療法)が標準となっています。しかし、ゲノム編集技術の急速な進歩により、将来的なパラダイムシフトの可能性が議論されています。
CRISPRゲノム編集治療への期待と「2つの大きな壁」
2025年5月、CRISPR-Cas9技術を用いたパーソナライズドゲノム編集治療がCPS1欠損症の新生児に対して世界初の臨床的成功を収め、希少疾患コミュニティに大きな希望を与えました。しかしASXL関連障害への応用には、2つの巨大な技術的障壁が立ちはだかっています[11]。
壁① 血液脳関門(BBB)の突破
CPS1欠損症は肝臓の疾患で、遺伝子編集ツールの送達技術がすでに確立されつつあります。一方ASXL3関連障害は病態の主座が脳(中枢神経系)にあり、厳密な関所である血液脳関門を超えて広範な神経細胞に編集ツールを安全に届ける技術は、神経科学・遺伝子治療分野における最大の未解決課題の一つです。
壁② 変異の多様性と遺伝子サイズ
ASXL3の病原性変異はエクソン11・12に無数に散在する多様なフレームシフトやナンセンス変異であり、全患者に適用可能な単一の編集ツールを設計することは困難です。さらに、正常な遺伝子を補充するアプローチでも、ASXL3遺伝子自体が巨大なため、送達に最適とされるAAVベクターの容量制限を超えてしまうという物理的な問題があります。
基礎科学への投資が「ミッシングリンク」を埋める
これらの障壁を迂回・克服するため、ARRE Foundationなどの研究推進組織は基礎科学への戦略的投資を最優先課題に据えています[12]。分子メカニズムが完全に解明されれば、巨大な遺伝子そのものを脳に届けるのではなく、PR-DUB複合体の下流で異常抑制されている遺伝子群のロックを解除する「低分子化合物」によるエピジェネティック治療薬という、全く新しい薬理学的アプローチが見えてきます。
日本国内でも、「先天異常症候群をもつ子と家族の支援システム(GENIE)」プラットフォームにおいてBainbridge-Ropers症候群が対象疾患として登録され、静岡県立こども病院・長野県立こども病院・東京都立小児総合医療センターなど高度遺伝診療を提供する中核施設が参加する形で、患者登録と自然歴収集を通じた支援体制が構築されつつあります[13]。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 ASXL3関連疾患・遺伝子検査のご相談
Bainbridge-Ropers症候群を含むASXL3関連障害や、
遺伝子検査・遺伝カウンセリングについて、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] NCBI Gene. ASXL3 ASXL transcriptional regulator 3 [Homo sapiens]. Gene ID: 80816. [NCBI Gene]
- [2] Schirwani S, et al. ASXL3-Related Disorder. GeneReviews® [Internet]. Seattle: University of Washington; 2020. [GeneReviews NBK563693]
- [3] SFARI Gene. ASXL3 — Gene Score and Evidence. Simons Foundation Autism Research Initiative. [SFARI Gene]
- [4] Woods E, et al. ASXL3-related disorder: molecular phenotyping and comprehensive review providing insights into disease mechanism. Clin Genet. 2024. [Clinical Genetics 2024]
- [5] Srivastava A, et al. De novo dominant ASXL3 mutations alter H2A deubiquitination and transcription in Bainbridge-Ropers syndrome. Hum Mol Genet. 2016;25(3):597-608. [PMC4731023]
- [6] Frontiers. ASXL3 gene variants causing Bainbridge-Ropers syndrome: clinical and genetic analysis of four Chinese patients. Front Neurosci. 2025. [Frontiers 2025]
- [7] Bainbridge MN, et al. De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome Med. 2013;5(2):11. [PMC3707024]
- [8] Paternal mosaicism in ASXL3-related Bainbridge-Ropers syndrome: implications for genetic counseling and prenatal diagnosis. Front Pediatr. 2025. [PMC12443740]
- [9] Modeling Bainbridge-Ropers Syndrome in Xenopus laevis Embryos. Front Physiol. 2020. [Frontiers Physiology 2020]
- [10] ASXL3 controls cortical neuron fate specification through extrinsic self-renewal pathways. bioRxiv. 2021. [bioRxiv 2021]
- [11] ARRE Foundation. A breakthrough in personalized gene editing—and what it means for ASXL-related disorders. [ARRE Foundation]
- [12] ARRE Foundation. A conversation with Dr. Karen Ho on the future of ASXL-related disorder research. [ARRE Foundation]
- [13] GENIE 希少疾患・未診断疾患支援システム. ASXL3 – Bainbridge-Ropers症候群. [GENIE]
- [14] OMIM. ASXL3 ASXL Transcriptional Regulator 3 Entry #615115. Johns Hopkins University. [OMIM #615115]
- [15] OMIM. Bainbridge-Ropers Syndrome Entry #615485. Johns Hopkins University. [OMIM #615485]






