目次

ANKRD11遺伝子は、ヒト染色体16番長腕末端(16q24.3)に位置する重要なクロマチン制御因子で、神経発達・骨格形成・転写ネットワーク全体を支える「指揮者」のような役割を担っています。この遺伝子の機能が失われると、知的障害・低身長・特徴的な顔貌・巨大歯を主徴とするKBG症候群の原因となります。2024〜2026年の研究により、コヒーシン複合体やSETD5遺伝子との分子連携が次々と解明されつつあり、希少疾患研究の最前線として国際的に注目されている遺伝子です。
Q. ANKRD11遺伝子とはどんな働きをしている遺伝子ですか?
A. 細胞核の中で「どの遺伝子をいつ・どのくらい使うか」を制御するクロマチン制御因子です。特に脳の神経細胞の発達と頭蓋顔面の骨形成において中心的な役割を果たし、機能が損なわれるとKBG症候群と呼ばれる発達障害・骨格異常を引き起こします。
- ➤基本情報 → 染色体16q24.3、17エクソン、OMIM 611192
- ➤分子機能 → HDAC3による転写抑制とp53の活性化、両方向の制御
- ➤最新研究 → コヒーシン複合体・SETD5軸の発見(2024〜2025年)
- ➤関連疾患 → KBG症候群(OMIM 148050)、常染色体顕性遺伝
- ➤検査・治療 → トリオWES/CMA/NIPT、2026年に専用成長チャートが公開
1. ANKRD11遺伝子とは:基本情報とゲノム上の位置
ANKRD11(Ankyrin Repeat Domain 11)遺伝子は、ヒトの第16番染色体長腕の末端付近、すなわち16q24.3と呼ばれる場所に位置している遺伝子です。17のエクソン(タンパク質の設計図となる部分)から構成されており、進化の過程で高度に保存されてきた「アンキリンリピートドメイン」という特徴的な構造を持つ巨大なタンパク質をコードしています。
| 項目 | 内容 |
|---|---|
| 遺伝子名 | ANKRD11(Ankyrin Repeat Domain 11) |
| 染色体位置 | 16q24.3 |
| エクソン数 | 17 |
| OMIM番号 | 611192(遺伝子)/148050(KBG症候群) |
| タンパク質機能 | クロマチン制御因子(転写の共役因子) |
| 主な発現組織 | 卵巣、脳、下垂体前葉、結合組織、肺など全身に広く |
| 関連疾患 | KBG症候群(常染色体顕性遺伝) |
ANKRD11は単一の臓器だけで働く遺伝子ではなく、生命維持や発生に欠かせない多くの組織で広く発現しています。特に卵巣・脳・下垂体前葉など内分泌系と中枢神経系で高い発現レベルを示し、上腕の腱、子宮内膜、皮膚、肺など、結合組織や末梢組織でも顕著な発現が確認されています。マウスを用いた研究でも、胚の神経発生領域や大動脈弁、視索上核、脊髄など、神経発生と心血管系の形成に関わる領域で強く働くことが報告されています。
💡 用語解説:常染色体顕性(優性)遺伝
「常染色体」とは性染色体(X・Y)以外の染色体を指し、「顕性(けんせい)」は以前「優性」と呼ばれていた言葉です。私たちは染色体を2本ずつ持っていますが、そのうち1本に変異があるだけで症状が出る遺伝形式が常染色体顕性遺伝です。ANKRD11関連疾患(KBG症候群)はこの遺伝形式をとります。ただし、報告されている多くのケースは両親に変異がなく、お子さんに初めて生じた「新生(de novo)変異」によるものです。
2. ANKRD11タンパク質の分子機能:クロマチン制御因子としての役割
ANKRD11タンパク質は細胞の核内に存在し、遺伝子の「読まれ方」を細やかに調節する司令塔として働きます。アンキリンリピートと呼ばれる構造を介してさまざまなタンパク質と物理的に結合し、転写を促進したり抑制したりする両方向の制御を行います。この多機能性こそが、ANKRD11が神経・骨格・内分泌など多臓器の発生に同時に関与できる理由です。
💡 用語解説:クロマチンとクロマチン制御因子
DNAは細胞の中で「ヒストン」というタンパク質に巻きついて折りたたまれており、この複合体をクロマチンと呼びます。クロマチンの「ほどけ具合」によって、その場所にある遺伝子が読まれやすくなったり読まれにくくなったりします。クロマチン制御因子とは、このほどけ具合をコントロールして遺伝子の発現を切り替える調節役のタンパク質群。ANKRD11はその代表選手のひとりで、必要なときに必要な遺伝子をオン・オフする「指揮者」のような役割を担っています。
HDAC3を呼び込んで転写を抑える「ブレーキ」機能
ANKRD11はアンキリンリピートを通じて、p160と呼ばれる共役因子や核内受容体複合体に結合します。そしてこの複合体に対して、ヒストン脱アセチル化酵素のひとつであるHDAC3を強力に呼び込みます。HDAC3が動員されると、標的となる遺伝子のプロモーター(遺伝子のスタート地点)周辺のクロマチンがぎゅっと折りたたまれ、結果として遺伝子が読まれにくくなる──つまり転写抑制が起こります。この働きから、ANKRD11は初期には乳がんなどの腫瘍を抑える「腫瘍抑制遺伝子」としても注目されてきました。
p53と協働して「アクセル」も踏む二面性
一方でANKRD11は、状況に応じて遺伝子発現を強力に「促進する」コアクチベーター(共役活性化因子)としても機能します。とくに重要なのが、細胞のストレス応答・DNA修復・アポトーシス(細胞死)の制御で中心的な役割を果たす転写因子p53との相互作用です。ANKRD11はp53と直接結合してp53のアセチル化を促し、p53のDNA結合能力と転写活性化能を大幅に強化します。さらに、p53とANKRD11の間には自己制御的なフィードバックループが形成されていることもわかっており、頭蓋顔面の発生プロセスにおける細胞の増殖と死の絶妙なバランスを支えています。

3. 最新研究:コヒーシン複合体・SETD5軸の発見(2024〜2025年)
長年にわたって、ANKRD11の機能不全がなぜKBG症候群のような複雑な表現型を引き起こすのか、その分子レベルでの全体像は完全には解明されていませんでした。しかし2024年から2025年にかけて、ANKRD11研究は決定的なパラダイムシフトを迎えました。コヒーシン複合体との直接結合と、SETD5遺伝子を介したタンパク質翻訳制御という、2つの新しい分子経路が次々に解明されたのです。
コヒーシン複合体との結合と染色体3D構造の制御
💡 用語解説:コヒーシン複合体
染色体の立体的な折りたたみを制御するリング状の巨大なタンパク質複合体です。離れた位置にある遺伝子の「スイッチ部分(エンハンサー)」と「読み取り開始部分(プロモーター)」を物理的に接触させ、必要な遺伝子発現を可能にします。コヒーシン関連遺伝子の異常はコルネリア・デ・ランゲ症候群(CdLS)の原因として古くから知られており、これらの疾患群は「コヒーシン病(Cohesinopathy)」と総称されます。
2024年に米国科学アカデミー紀要(PNAS)に掲載された画期的な研究によって、ANKRD11がコヒーシン複合体と直接結合することが初めて証明されました。高解像度の結晶構造解析(3.2オングストロームの分解能)により、ANKRD11はその拡張された「YEFモチーフ」を含む特定のフラグメント(アミノ酸残基342〜378)を介して、コヒーシン複合体の主要構成要素であるSTAG2-RAD21サブ複合体に対し、極めて高い親和性と特異性で結合することが明らかになりました。さらに重要なことに、ANKRD11は染色体構造の構築因子CTCFと、コヒーシン結合を巡って競合する能力を持っており、これによって細胞系統ごとに固有の遺伝子発現の3D構造をダイナミックに形成しています。
この発見の生理的意義を検証するため、研究グループはコヒーシン結合だけを特異的に壊すY347A変異を導入したマウスを作出。このマウスは、前頭骨の喪失や自閉症様行動など、ヒトのKBG症候群患者に酷似した深刻な神経・頭蓋顔面異常を引き起こしました。これは、KBG症候群が広い意味で「コヒーシン病」のひとつであり、コルネリア・デ・ランゲ症候群と共通の分子基盤を持つことを論理的に示した画期的な成果といえます。
ANKRD11-SETD5軸:rRNA合成とタンパク質翻訳の制御
💡 用語解説:rRNA(リボソームRNA)と翻訳
細胞がタンパク質をつくる工場が「リボソーム」で、その骨格を構成するRNAがrRNA(リボソームRNA)です。リボソームでmRNAの情報がタンパク質へと「翻訳」されます。神経細胞のように活発に働く細胞ほど大量のタンパク質を必要とするため、rRNAの合成量が細胞の活動レベルを大きく左右します。神経発達障害とリボソーム関連遺伝子の異常は密接にリンクしていることが近年わかってきました。
2025年に発表された細胞生物学研究では、ANKRD11が別の強力な知的障害関連遺伝子SETD5と階層構造(ANKRD11-SETD5軸)を形成していることが明らかになりました。ANKRD11はSETD5遺伝子のプロモーター領域に結合し、ヒストンH3K4のメチル化を担う複合体の構成要素であるWDR5をリクルートします。これによりSETD5プロモーター上のH3K4トリメチル化(H3K4me3)が促進され、SETD5の発現が強力に活性化されるという仕組みです。
SETD5はrRNAの転写活性化因子として機能する酵素ですから、ANKRD11はSETD5を介して細胞全体のタンパク質合成能力に間接的に影響を及ぼします。実際に、ANKRD11が不足した神経モデル細胞では、成熟28S rRNAやpre-rRNAのレベルが顕著に低下し、全体的なタンパク質翻訳能の減衰と細胞増殖の阻害が観察されました。注目すべきは、ANKRD11欠損細胞に外因的にANKRD11またはSETD5のいずれかを過剰発現させると、低下していたrRNAレベルと翻訳活性が完全に回復したことです。この「レスキュー」の成功は、SETD5や翻訳経路をターゲットにした薬剤がKBG症候群の新たな治療候補となる可能性を強く示唆しています。
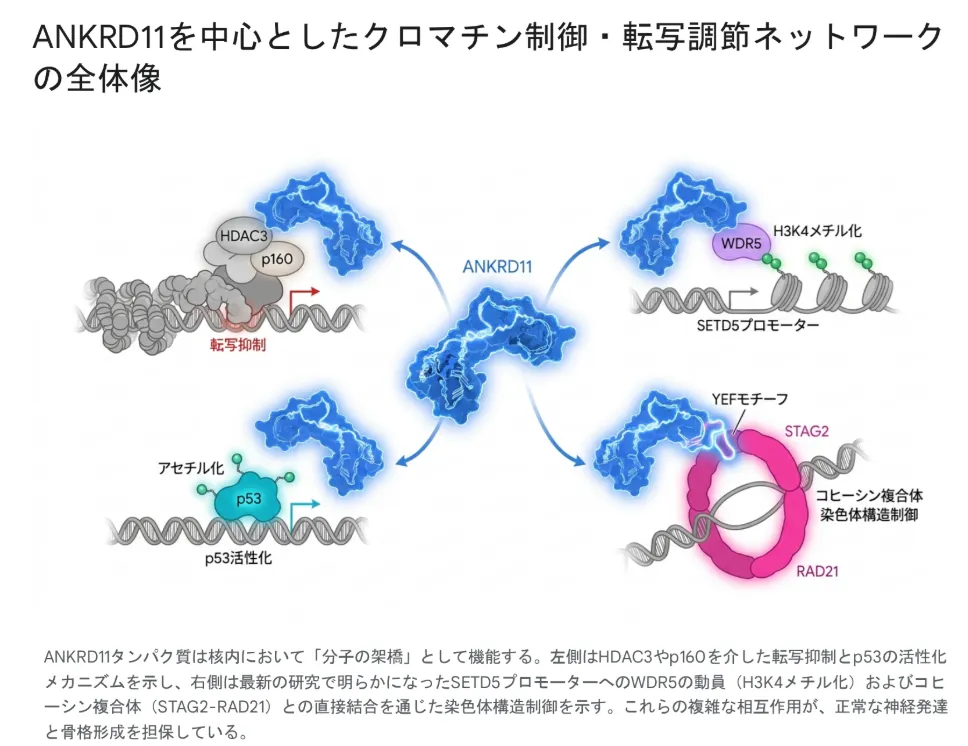
ANKRD11タンパク質は核内で「分子の架橋」として働く。左側はHDAC3/p160を介した転写抑制(左下)とp53のアセチル化を介した活性化(左上)という古典的メカニズム、右側は2024〜2025年の研究で明らかになったSETD5プロモーターへのWDR5動員によるH3K4メチル化(右上)とコヒーシン複合体STAG2-RAD21との直接結合による染色体3D構造制御(右下)を示す。これら4方向の相互作用が同時に働くことで、正常な神経発達と頭蓋顔面の骨格形成が支えられている。
4. 発生過程におけるANKRD11の役割:頭蓋顔面と神経発達
ANKRD11が個体レベルでどのような働きをしているのかを解明するため、世界中でさまざまなマウスモデルが開発されてきました。完全にホモ接合体(両方の遺伝子が壊れた状態)のマウスは胎齢9日という極めて早い時期に死亡してしまうため、特定の組織だけでANKRD11を欠損させる「条件付きノックアウト(CKO)」モデルが用いられています。とくに頭蓋顔面の形成を担う神経堤細胞に絞った研究が、KBG症候群の病態解明に大きく貢献しました。
💡 用語解説:神経堤細胞(しんけいていさいぼう)
胚発生のごく初期に神経管の縁から離れて、全身のあちこちに移動していく特殊な細胞集団です。顔面の骨や軟骨、末梢神経、色素細胞、心臓の一部など、極めて多様な組織のもとになります。神経堤細胞の発生異常は「神経堤症」と総称され、CHARGE症候群、トリーチャー・コリンズ症候群、ワーデンブルグ症候群など多くの先天性疾患の背景にあります。KBG症候群の頭蓋顔面異常も、神経堤細胞でANKRD11が正しく働かないことが根本原因と考えられています。
マウスモデルが示した骨化中心と口蓋発生の異常
神経堤細胞特異的なANKRD11ホモ接合体欠損マウスは、出生時に致死となるほどの重篤な発生異常を示します。具体的には、口蓋裂・小顎症(後退顎)・重度の中顔面低形成・頭蓋冠の成長低下といった、KBG症候群の臨床像を重症化したような所見が観察されます。ヘテロ接合体(片方だけ欠損)マウスはより軽度の中顔面低形成や、頭蓋骨の癒合遅延による持続的な大泉門の開存を示し、これはヒトKBG症候群患者の特徴を忠実に再現しています。
詳細な発生学的観察から、ANKRD11は骨化中心の「立ち上がり」自体には影響しないものの、その後の「拡張と癒合」のプロセスに必須であることがわかってきました。骨形成のマスター転写因子であるRunx2やSp7(Osterix)の発現ドメインがANKRD11の欠損で顕著に変化することから、ANKRD11は骨化の遺伝子スイッチを適切なタイミングでオンにする役割を担っていると考えられます。胎齢13.5日の口蓋棚における局所的な細胞増殖もANKRD11がなければ低下し、口蓋裂の形成へとつながります。
5. ANKRD11の機能不全が引き起こすKBG症候群
ANKRD11遺伝子の機能喪失型変異や、16q24.3領域を含む微小欠失は、KBG症候群(OMIM 148050)の直接的な原因となります。1975年にHerrmannらが最初に記述した3家系の患者のイニシャル(K・B・G)にちなんで名付けられた、常染色体顕性遺伝形式をとる希少疾患です。
💡 用語解説:ハプロ不全(Haploinsufficiency)
遺伝子は通常、父親由来と母親由来の2コピーが存在します。ハプロ不全とは、片方のコピーが機能を失うことで、残った1コピーだけでは細胞が必要とする量のタンパク質を供給できなくなる状態のこと。ANKRD11ではこのハプロ不全がKBG症候群の主要な発症メカニズムで、報告されているANKRD11変異の約91%はナンセンス変異やフレームシフト変異など、タンパク質を作れなくする「機能喪失型」変異です。
KBG症候群の主な特徴(概要)
🦷 歯科・顔貌
- 上顎中切歯の巨大歯:85〜95%
- 三角顔、癒合眉、低い生え際
- 突出した鼻梁、長い人中
🧠 神経発達
- 発達遅滞:90%以上
- 知的障害(多くは軽度)
- てんかん発作、EEG異常
📏 成長・骨格
- 出生後の低身長:54%
- 何らかの骨格異常:92%
- 手の異常・脊椎異常
👂 その他
- 難聴(伝音・感音・混合):約27%
- 哺乳・摂食障害
- 先天性心疾患(一部)
2026年の新展開:疾患特異的成長チャートと成長ホルモン療法
KBG症候群の低身長治療として、組換えヒト成長ホルモン(rhGH)療法が長らく検討されてきましたが、これまでは小規模な症例報告にとどまっていました。2026年にLow KJらが発表したA Growth Chart for KBG Syndrome研究により、この状況は大きく変わりました。28名の追跡データから、治療開始前の身長SDS(標準偏差スコア)の中央値は−2.9と深刻な低身長状態でしたが、治療後は−1.8にまで劇的に改善。治療を受けた患者の約半数が1 SDS以上の身長ゲインを獲得し、治療前に「低身長」とされていた18名のうち10名が正常範囲内に到達しました。
この研究で特筆すべきは、治療開始前に明確な成長ホルモン分泌不全(GHD)の基準を満たしていた患者と、満たしていなかった患者で、SDSの改善幅にほとんど差がなかったという点です。これは、厳密な分泌不全が証明されないKBG症候群の低身長に対しても、rhGH療法が十分な治療選択肢となりうることを支持する画期的な知見です。同研究グループはLMSz法という統計手法でKBG症候群専用の成長チャートも世界で初めて構築し、健常児の標準曲線では把握しきれなかった患者の成長パターンを正確にモニタリングできるようになりました。
6. ANKRD11遺伝子に関連する検査
ANKRD11の変異は、コーディング領域の微細な変化から16q24.3領域全体を巻き込む微小欠失まで、多様なパターンをとります。そのため診断には複数の検査法を組み合わせた多角的アプローチが現在の標準です。永久歯の巨大歯が萌出する前の乳幼児期は顔貌特徴も非特異的なため、診断には遺伝子レベルの解析が決定的な役割を果たします。
💡 用語解説:トリオ全エクソーム解析(Trio-WES)
遺伝子の中でタンパク質をコードする領域(エクソン)全体を網羅的に解析する次世代シーケンス手法をWESと呼びます。「トリオ」とは、お子さん本人と両親の合計3名分を同時に解析する方式。両親には存在せずお子さんに新たに生じた新生(de novo)変異を高精度に検出できるため、KBG症候群のように多くが新生変異で発症する疾患の診断に有用です。
💡 用語解説:マイクロアレイ染色体検査(CMA)
染色体上の微小な欠失や重複を網羅的に検出する検査です。一般的な顕微鏡でのGバンド法では5Mb(メガベース)以下の欠失は見つけられませんが、CMAは数十kb程度の微小欠失まで検出可能。16q24.3を含む微小欠失型のKBG症候群(連続遺伝子症候群)の診断にはCMAが第一選択となります。染色体検査(Gバンド法)と組み合わせて行うこともあります。
ANKRD11が対象に含まれる主な検査メニュー
| 検査メニュー | 主な対象と特徴 |
|---|---|
| NIPTインペリアルプラン | 154遺伝子218疾患をカバーする出生前検査。ANKRD11も対象遺伝子に含まれます。 |
| 発達障害・学習障害・知的障害遺伝子パネル | 563遺伝子を網羅的に解析。出生後の発達遅滞や知的障害の原因精査に。 |
| 染色体シーケンス解析(CSA) | NGSベースのコピー数解析とシーケンス解析を統合した検査。 |
| 羊水検査・絨毛検査+CMA | 出生前の確定診断。胎児超音波で構造異常がある場合などが対象。 |
なお、2022年以降の分子診断学の進展により、WESやCMAでも原因が特定できなかった定型的なKBG症候群患者に対して、RT-qPCRによるANKRD11 mRNA発現アッセイを行う手法が確立されつつあります。コーディング領域の異常ではなく、プロモーターやイントロンといった「制御領域」に隠れた病原性変異が見つかるケースも報告されており、診断率の継続的な向上に貢献しています。また、Face2GeneなどのAI支援フェノタイピングツールが、特に乳幼児期の特徴的な顔貌パターンを客観的に評価する補助手段として臨床現場に導入されつつあります。
7. 遺伝カウンセリングの意義
ANKRD11関連疾患(KBG症候群)の診断前後に、専門医による遺伝カウンセリングを受けることには大きな意義があります。臨床遺伝専門医はご家族の状況を多角的に評価し、検査の選択肢や将来の見通しについて中立的な立場で情報を提供します。
- ➤遺伝形式と再発リスクの説明:多くのケースは新生(de novo)変異であり、ご両親には変異がないことがほとんどです。ただし常染色体顕性遺伝のため、患者本人がお子さんを持つ場合の遺伝確率は理論上50%です。生殖細胞モザイクの可能性も視野に入れた説明が行われます。
- ➤表現型の幅についての情報提供:KBG症候群は同じ家系内でも重症度に大きなばらつきがあります。知的障害の程度や成長への影響は患者ごとに異なり、画一的な予測は困難です。
- ➤出生前診断の選択肢:家族内に既知の変異がある場合は、絨毛検査・羊水検査による出生前遺伝子診断が技術的に可能です。検査を受けるか受けないかの決定は、ご家族の価値観を最優先にして慎重に行われるべきものです。
- ➤長期的サポートの設計:診断確定後の発達支援・教育計画・成長ホルモン療法の適応評価など、ライフステージに応じた包括的な医療連携が重要です。遺伝カウンセリングについて詳しく
8. よくある誤解
誤解①「ANKRD11は1つの臓器だけの遺伝子」
ANKRD11は脳・骨・心臓・卵巣・肺など全身の組織で広く発現するクロマチン制御因子です。だからこそ、機能が損なわれると複数の臓器が同時に影響を受けるのです。
誤解②「両親が健康なら遺伝ではない」
KBG症候群の多くは新生(de novo)変異によって発症します。ご両親には変異がなく、お子さんに初めて生じた変異であることがほとんどです。「遺伝ではない」と即断して診断が遅れることを防ぎたいケースです。
誤解③「巨大歯がなければKBGではない」
上顎中切歯の巨大歯は85〜95%と高頻度ですが、永久歯が萌出する前の乳幼児期には観察できません。発達遅滞・低身長・特異な顔貌など他のサインから疑い、遺伝子検査で確定するアプローチが大切です。
誤解④「成長は背が低いまま諦めるしかない」
2026年の大規模研究で、成長ホルモン療法(rhGH)の有効性がエビデンスとして確立されました。治療を受けた患者の過半数が低身長から正常範囲内に到達したという報告があり、内分泌科との連携が重要です。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 ANKRD11関連疾患・希少遺伝子疾患のご相談
ANKRD11・KBG症候群をはじめとする希少遺伝性疾患のご相談は、
臨床遺伝専門医が在籍するミネルバクリニックへお気軽にどうぞ。
関連記事
参考文献
- [1] NCBI Gene. ANKRD11 ankyrin repeat domain 11 [Homo sapiens (human)]. Gene ID: 29123. [NCBI Gene]
- [2] OMIM. ANKRD11 (*611192) / KBG Syndrome (#148050). Johns Hopkins University. [OMIM 611192] / [OMIM 148050]
- [3] Morel Swols D, Foster J 2nd, Tekin M. KBG Syndrome. In: GeneReviews®. University of Washington, Seattle. [GeneReviews NBK487886]
- [4] Orphanet. KBG syndrome. ORPHA:2332. [Orphanet ORPHA:2332]
- [5] Liu J, et al. ANKRD11 binding to cohesin suggests a connection between KBG syndrome and Cornelia de Lange syndrome. PNAS. 2025. [PNAS 2025]
- [6] KBG syndrome-associated protein ANKRD11 regulates SETD5 expression to modulate rRNA levels and translation. PMC. 2025. [PMC12164204]
- [7] Goodkey K, et al. The Chromatin Regulator Ankrd11 Controls Palate and Cranial Bone Development. Front Cell Dev Biol. 2021. [PMC8117352]
- [8] Insights into the ANKRD11 variants and short-stature phenotype through literature review and ClinVar database search. PMC. 2024. [PMC11318275]
- [9] Low KJ, et al. The LMSz method – an automatable scalable approach to constructing gene-specific growth charts in rare disorders. ResearchGate. 2026. [ResearchGate]
- [10] GeneCards. ANKRD11 Gene – Ankyrin Repeat Domain 11. [GeneCards]






