目次

KBG症候群は、ANKRD11遺伝子(16番染色体長腕24.3)のヘテロ接合性変異によって生じる、世界で340名以上が報告されている希少な常染色体顕性(優性)遺伝疾患です。上顎中心切歯の巨歯症、三角形の顔立ち、低身長、発達の遅れを4本柱とし、近年は成人期の大動脈拡張や強迫性障害(OCD)の高頻度など、生涯にわたって多臓器に影響する進行性疾患であることが明らかになっています。
Q. KBG症候群とはどのような疾患ですか?まず結論だけ知りたいです
A. ANKRD11遺伝子の機能喪失によって生じる、特徴的な顔貌・上顎中心切歯の巨歯症・低身長・発達遅滞を主徴とする常染色体顕性遺伝疾患です。1975年にHerrmannらによって命名され、世界で340名以上の患者さんが報告されています。成人期にも大動脈拡張や強迫性障害など重要な合併症が続くため、生涯にわたる集学的管理が必要です。
- ➤疾患の定義 → OMIM 148050、ANKRD11遺伝子変異、世界340名以上の報告
- ➤分子メカニズム → ハプロ不全とコヒーシン複合体結合障害による「クロマチノパチー」
- ➤主な症状 → 巨歯症・特徴的顔貌・低身長・骨年齢遅延・発達遅滞
- ➤精神・行動面 → 強迫性障害82%・ADHD29%という独特な精神医学的プロファイル
- ➤最新治療 → メチルフェニデートの有効性が臨床試験で実証
1. KBG症候群とは:疾患の定義と歴史的背景
KBG症候群(OMIM 148050)は、1975年にアメリカの遺伝学者Herrmannらによって初めて医学文献に報告された希少な遺伝性疾患です。「KBG」という名称は、最初に報告された3家系の患者さんの名字の頭文字(K・B・G)から取られています。
本疾患は、特徴的な顔貌・上顎中心切歯の巨歯症・低身長と骨格異常・神経発達遅滞という4本柱で特徴づけられます。これらの所見は乳児期にはまだはっきりせず、永久歯が生え変わる7〜8歳頃以降に揃ってくるため、学童期になってから診断されるケースが少なくありません。
💡 用語解説:常染色体顕性(優性)遺伝
「常染色体」とは、性染色体(X・Y)以外の染色体のこと。「顕性(旧名:優性)」とは、2本ある遺伝子のうち片方に変異があるだけで症状が現れる遺伝形式です。KBG症候群では、変異した遺伝子を1つ持つだけで発症します。理論上、罹患した親から子へ伝わる確率は50%です。ただしKBG症候群の多くは、両親には変異がなく子どもで初めて生じた新生突然変異(de novo変異)として発症するため、実際には親子間で受け継がれるケースは少ないのが現状です。
2026年現在、世界で少なくとも340名以上の患者さんが医学文献に記述されており、次世代シーケンサー(NGS)や全エクソーム解析の普及により、確定診断例は加速度的に増加しています。軽症例や非典型例の見落としが多いことから、実際の有病率は報告数を大幅に上回ると推測されています。
歴史的にKBG症候群は「小児期の奇形症候群」として認識されてきましたが、近年の大規模成人コホート研究によって疾患の捉え方が大きく変わりました。2024年には91名の成人患者さん(16〜86歳)を対象とした国際的な調査が発表され、成人期にも大動脈拡張・骨粗鬆症・慢性便秘・睡眠障害・強迫性障害などの合併症が続くことが明らかになっています。現在では、KBG症候群は生涯にわたって多臓器システムに影響を及ぼす進行性かつ多面的な疾患として理解されています。
2. 原因遺伝子ANKRD11と分子病態メカニズム
KBG症候群の原因は、16番染色体長腕(16q24.3)に位置するANKRD11遺伝子のヘテロ接合性病的変異、または同遺伝子を含む16q24.3領域の微小欠失です。臨床的に同定される変異の約76%はANKRD11遺伝子そのものの点変異・小欠失で、残り約24%は16q24.3微小欠失です。
💡 用語解説:ANKRD11遺伝子とは
ANKRD11(Ankyrin Repeat Domain-containing Protein 11)は、全身の細胞で広く発現する転写因子をコードする遺伝子です。1つの転写活性化ドメインと2つの転写抑制ドメインを持ち、クロマチン(DNAとタンパク質の複合体)の状態を細やかに調整する「クロマチンリモデリング・コレギュレーター」として機能します。神経の発達や骨を作る細胞の働きに欠かせない遺伝子発現プログラムを制御しており、特に胎児期の脳形成と頭蓋顔面・骨格発生において重要な役割を担います。
ハプロ不全がKBG症候群の出発点
KBG症候群で同定される変異の大多数は、タンパク質が途中で切断されてしまうフレームシフト変異やナンセンス変異による機能喪失型変異です。この結果、本来2本ある遺伝子のうち1本が機能を失い、必要なANKRD11タンパク質量が半分しか作られないハプロ不全という状態が生じます。
💡 用語解説:ハプロ不全・ミスセンス変異・新生突然変異
ハプロ不全(haploinsufficiency)とは、2本ある遺伝子のうち1本が壊れたことで、タンパク質の量が半分に減って症状が出る状態です。「半分では足りない」という意味で、KBG症候群の主要メカニズムです。
ミスセンス変異は、DNAの塩基が1つ変化することでアミノ酸が別の種類に置き換わるタイプの変異で、タンパク質の形と機能を変化させます。
新生突然変異(de novo変異)とは、両親の生殖細胞(精子・卵子)または受精直後に新たに生じた変異で、両親には同じ変異がない状態を指します。KBG症候群では病的変異の約3分の2が新生突然変異です。
最新発見:コヒーシン複合体との結合とクロマチノパチー
2024年に米国科学アカデミー紀要(PNAS)に発表された画期的な研究によって、ANKRD11タンパク質が染色体の三次元構造を制御する「コヒーシン複合体」のSTAG2-RAD21サブコンプレックスと直接結合することが証明されました。ANKRD11内の「YEFモチーフ」を含むフラグメントが、コヒーシンに対して高い親和性と特異性で結合し、神経発生関連遺伝子(Ece1・Ephb2など)や骨発達関連遺伝子(Matn1・Msx2・Bmp7など)の転写を協調的に制御していることが明らかになっています。
💡 用語解説:コヒーシン複合体とクロマチノパチー
コヒーシン複合体は、染色体を糸のように引き寄せて立体的なループ構造を作り出す分子マシンです。これによってDNA上の特定の遺伝子が、必要なタイミングで「オン/オフ」されるよう調節されます。胚発生や細胞分裂に欠かせない役割を担います。
クロマチノパチー(chromatinopathy)とは、クロマチン(DNAの折りたたみ構造)を制御する遺伝子の異常によって生じる疾患群の総称です。KBG症候群は、ANKRD11とコヒーシンの結合不全により、コルネリア・デ・ランゲ症候群と分子病態レベルで深く関連する「コヒーシノパチー」のスペクトラムに位置づけられるようになりました。
マウスモデルでANKRD11とコヒーシンの結合だけを特異的に妨げる変異(Tyr347→Ala)を導入すると、前頭骨の欠損や自閉症様の行動異常など、ヒトのKBG症候群と一致する表現型が再現されます。これらの結果は、ANKRD11とコヒーシンの相互作用が、KBG症候群の頭蓋顔面・骨格・神経の発達異常の分子レベルの起点であることを示しています。
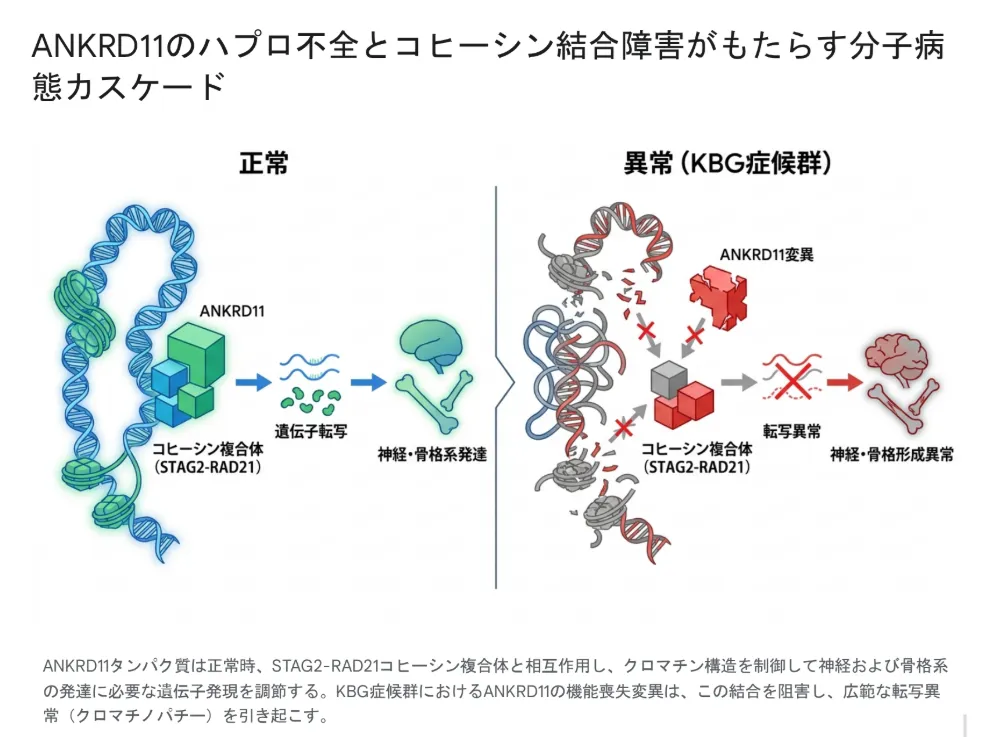
ANKRD11タンパク質は正常時、STAG2-RAD21コヒーシン複合体と結合してクロマチン構造を制御し、神経・骨格系の発達に必要な遺伝子発現を調節する。KBG症候群におけるANKRD11の機能喪失変異はこの結合を阻害し、広範な転写異常(クロマチノパチー)を引き起こす。
遺伝子型と表現型の相関、腫瘍リスクの可能性
短縮型変異(フレームシフトやナンセンス変異)を持つ患者さんは、ミスセンス変異の方と比較して、低身長や骨格奇形・全般的な発達遅滞・知的障害・学習困難の発現頻度が統計学的に有意に高い(p<0.05)と報告されています。また、ANKRD11単独の変異ではなく16q24.3微小欠失を持つ症例では、自閉症スペクトラム障害の重症化や脳構造異常が加わり、独立した「16q24.3微小欠失症候群」として扱われることもあります。
腫瘍学的観点では、ANKRD11遺伝子座全体を喪失した17歳の患者さんに傍精巣性悪性ラブドイド腫瘍が発症した症例が報告されており、ANKRD11が腫瘍抑制遺伝子として機能している可能性が指摘されています。普遍的な癌スクリーニングを推奨するほどのデータはまだありませんが、長期フォローアップの中でこの潜在的リスクを念頭に置く重要性が認識され始めています。
3. 主な症状と臨床的特徴
KBG症候群の臨床像は、年齢・性別・同じ家系内であっても重症度に大きなばらつきがあります。発達段階に応じて症状の見え方が変化する「動的な疾患モデル」として理解する必要があります。
🦷 歯科・顔貌
- 上顎中心切歯の巨歯症(最重要所見)
- 三角形の顔立ち・短頭症
- 癒合眉・両眼開離
- 突出した鼻梁・前向き鼻孔
- 長い人中・薄い上唇
📏 骨格・成長
- 低身長(10パーセンタイル未満)
- 骨年齢の遅延(2SD以上)
- 短指症・第5指弯曲
- 大泉門の閉鎖遅延(約22%)
- 脊柱側弯症・肋骨椎骨異常
🧠 神経・発達
- 運動発達の遅滞
- 発語の遅滞
- 軽度〜中等度の知的障害
- てんかん(20〜40%)
- 脊髄係留症候群(約11%)
❤️ 全身・その他
- 先天性心疾患(10〜26%)
- 難聴(25〜31%・反復性中耳炎)
- 視覚異常(乱視・近視・斜視)
- 男児の停留精巣
- 口蓋裂・高口蓋
💡 用語解説:巨歯症(macrodontia)
上顎の中心切歯(一番真ん中の前歯)が著しく大きいことを指します。KBG症候群の最も特異的な所見で、診断基準では永久歯の中心切歯の幅が男性で10mm以上、女性で9.7mm以上あることが要件とされています。乳歯では現れず、6〜7歳頃に永久歯が生え変わるタイミングで気づかれることがほとんどです。歯科医師から「前歯がやけに大きい」と指摘されて遺伝相談につながるケースも少なくありません。
新生児期に注意すべき重大な合併症
新生児期にはKBG症候群らしい特徴がまだ揃わないため、診断が難しい時期ですが、いくつかの重要なサインがあります。在胎不当過小(SGA)として出生する割合は41.1%、新生児期の哺乳障害は約52.1%に見られます。哺乳困難が重症な場合は経鼻胃管栄養や胃瘻造設が必要になることもあります。
さらに近年、極めて特異かつ重大な新生児合併症として、先天性乳び胸(congenital chylothorax)を伴う症例が複数報告されています。乳び胸はリンパ液が胸腔内に貯留する致死的な呼吸器合併症で、ANKRD11の機能不全が胎生期のリンパ管ネットワーク発達に影響する可能性が示唆されています。原因不明の先天性乳び胸や胸水貯留を有する新生児では、KBG症候群を早期鑑別診断の一つとして考慮する重要性が高まっています。
特異的な精神・行動プロファイル
KBG症候群の精神医学的特徴は、単なる「知的な遅れ」では片付けられない独特なプロファイルを持ちます。患者さんや介護者のQOL(生活の質)を最も大きく左右する要因は、しばしば身体的奇形よりもこれらの精神医学的合併症であることが、近年の研究で明らかになっています。
特に注目すべきは、強迫性障害またはそれに準じた強迫的特性が約82%という驚異的な高頻度で見られる点です。一般集団のOCD有病率(約7.1%)と比べて10倍以上であり、しかも症状の重症度は患者さんの認知機能レベル(IQ)とは独立しています。これは、ANKRD11の変異が大脳基底核や前頭葉ネットワークに直接的な影響を及ぼしている可能性を示唆する重要な所見です。
一方で、KBG症候群の患者さんは典型的な自閉症のような完全な社会的孤立を好むわけではなく、むしろ社交的で愛情深く、ユーモアのセンスがあるという性格特性を持つ方が多いと報告されています。社会的な空気を読む力の弱さや強いこだわり、過度な不安が対人関係で誤解を生むという、独特なパラドックスを抱えています。
4. 鑑別診断:類似する症候群との違い
KBG症候群の診断における最大の難しさは、「年齢依存的な表現型の進化」と「他症候群との表現型のオーバーラップ」にあります。乳幼児期には特徴的所見が揃わないため、別の遺伝性疾患と誤診されるリスクが高いのです。
コルネリア・デ・ランゲ症候群との鑑別
共通点:NIPBLやRAD21遺伝子の変異によるコヒーシノパチーであり、KBG症候群と分子レベルで関連。発達遅滞・低身長・特徴的顔貌など重なる症状が多い。
鑑別ポイント:巨歯症の有無、特徴的顔貌の形質、原因遺伝子の同定(ANKRD11 vs NIPBL/RAD21)。乳児期にはコルネリア・デ・ランゲ症候群と誤診されるケースが報告されている。
コフィン・シリス症候群との鑑別
共通点:知的障害・粗野な顔貌・骨格異常などの組み合わせから初期に鑑別診断に挙がる。
鑑別ポイント:コフィン・シリス症候群はARID1B等のSWI/SNF複合体関連遺伝子の変異が原因。第5指爪・指の低形成が特徴。遺伝子検査で明確に鑑別される。
ニコライデス・バライツァー症候群との鑑別
共通点:発達遅滞・粗野な顔貌・骨格異常。
鑑別ポイント:SMARCA2遺伝子変異が原因で、進行性の毛髪の希薄化・指の関節腫大・てんかんが特徴。KBGの巨歯症・癒合眉などの所見が決め手となる。
日本人を含むアジア人での注意点:韓国・日本・中国などのアジア人集団では、欧米例と比較してKBG症候群の顔貌の特徴がより微妙(subtle)に表現される傾向があり、見落としやすいことが指摘されています(Ascertainment bias)。単独の臨床所見に依存せず、原因不明の発達遅滞や低身長を有する場合はANKRD11を含む網羅的遺伝子パネル検査を積極的に検討することが推奨されています。
5. 診断基準と遺伝子検査の進め方
世界的に統一された診断基準はまだ確立されていませんが、現在最も広く支持されているのは2016年にLowらによって提唱された診断アルゴリズムです。
Low et al. 2016 臨床診断基準
発達遅滞・認知機能障害・著しい行動上の問題を有する発端者において、以下を満たす場合にKBG症候群が強く疑われます。
📋 KBG症候群が強く疑われる組み合わせ
- ➤主要基準を2つ以上満たす場合
- ➤主要基準1つ+その他の基準を2つ以上満たす場合
| 分類 | 所見の詳細 |
|---|---|
| 主要基準 | ① 巨歯症(永久歯上顎中心切歯の幅:男性10mm以上、女性9.7mm以上) ② 特異的顔貌(三角形の顔・癒合眉・両眼開離・前向き鼻孔・薄い上唇など) ③ 出生後の低身長(同年齢の10パーセンタイル未満) ④ 骨年齢の遅延(暦年齢の平均より2SD以上) |
| その他の基準 | ⑤ 骨格異常(短指症・第5指弯曲・大泉門閉鎖遅延・脊柱側弯症など) ⑥ 聴覚障害(伝音性・感音性・混合性難聴、反復性中耳炎) ⑦ 口蓋・毛髪・その他の異常(高口蓋・粗い毛髪・停留精巣など) |
出生後の確定診断:遺伝子検査が必須
最終的な確定診断は、全エクソームシーケンス(WES)などの次世代シーケンス技術、または染色体マイクロアレイ検査(CMA)を用いて、ANKRD11遺伝子における病的変異の存在、あるいは16q24.3領域の微小欠失を同定することで行われます。
従来のGバンド法(通常の染色体検査)では16q24.3の微小欠失は検出できません。原因不明の発達遅滞があり、特徴的所見が揃わない場合でも、染色体マイクロアレイや遺伝子パネル検査の適応を積極的に検討する必要があります。
出生前診断について:慎重な遺伝カウンセリングが前提
KBG症候群は表現型が広く、症状の重症度に大きなばらつきがある疾患です。出生前に変異が見つかったとしても、出生後の症状がどの程度になるかを正確に予測することは現時点では困難です。そのため、出生前診断を「常に受けるべきもの」として扱うのではなく、ご家族の価値観・人生観をふまえた丁寧な遺伝カウンセリングを経たうえで、ご家族自身がお決めになる事柄です。
既に家系内で病的変異が同定されている場合は、絨毛検査(CVS)または羊水検査+染色体マイクロアレイ検査または標的シーケンスによる胎児の確定診断が技術的には可能です。新生突然変異が大半を占めるKBG症候群では、第1子の出生前診断で変異を検出するのは現実的には難しく、家族歴がない場合の出生前スクリーニングは限定的です。

6. 治療と生涯にわたる集学的管理
KBG症候群は単一の専門科では対応できず、小児科・臨床遺伝科・整形外科・内分泌科・神経内科・循環器内科・心理士・歯科医などが連携する多職種チーム医療が不可欠です。年齢に応じて重点的なフォローアップ項目が変化していきます。
| 臓器系 | 主な合併症 | 推奨される管理 |
|---|---|---|
| 神経系 | てんかん発作(20〜40%)、脊髄係留症候群(約11%) | 診断時のベースラインEEG実施、抗てんかん薬による標準治療、仙骨部皮膚陥凹がある場合はMRIで脊髄係留を評価 |
| 循環器系 | 先天性心疾患(10〜26%、VSD・ASDなど)、成人期の大動脈基部拡大 | 診断時の心電図・経胸壁心エコー、定期的な大動脈径モニタリング、拡張時はβ遮断薬等を検討 |
| 感覚器 | 難聴(25〜31%)、反復性滲出性中耳炎、乱視・近視・斜視 | 5歳頃までの定期聴力検査、必要に応じて鼓膜換気チューブ留置、定期眼科健診 |
| 内分泌・骨格 | 低身長・骨年齢遅延、思春期早発、成人期の骨粗鬆症 | 成長速度のモニタリング、思春期早発症にはGnRHアナログ療法、骨密度の長期評価 |
| 消化器(成人期) | 慢性便秘(29%)、胃食道逆流症(21%)、腹部片頭痛、周期性嘔吐症候群 | 成人期も継続的な消化器症状の評価と症状管理 |
特に注意すべき大動脈基部拡大
成人期にかけて進行する大動脈基部拡大は、生命予後に直結する重要な合併症です。大動脈解離を引き起こすリスクがあるため、定期的な心エコー・CT・MRIによる画像監視が推奨されます。拡張が確認された場合、米国心臓病学会(ACC)・米国心臓協会(AHA)の遺伝性大動脈疾患ガイドラインに準拠し、β遮断薬またはアンジオテンシン受容体拮抗薬(ARB)の最大忍容用量での薬物療法を速やかに開始します。大動脈基部径が5.0cmに達した場合、または4.5cmでも家族歴や急速な拡張など高リスク因子がある場合は、予防的な大動脈基部置換術の適応を検討します。
最新治療:メチルフェニデートのN-of-1臨床試験
KBG症候群に伴うADHD症状に対しては、メチルフェニデート塩酸塩の有効性を検証する画期的な臨床試験(NCT06465641)が実施されました。6〜20歳の患者さんを対象としたプラセボ対照クロスオーバー中止試験(N-of-1試験)で、プラセボ投与期と比較してメチルフェニデート投与期では、教師による評価で多動性・衝動性が統計学的に有意に低下することが確認されました(p=0.04)。試験終了後、対象患者の約3分の2が治療継続を選択しています。一般集団のADHDに用いられる中枢神経刺激薬がKBG症候群の患者さんでも有効かつ安全な選択肢となり得ることが、ようやくエビデンスとして示された意義は大きいといえます。
7. 遺伝カウンセリングの意義
KBG症候群の確定診断後、ご家族への丁寧な遺伝カウンセリングは、医療管理と並んで欠かせない柱です。臨床遺伝専門医または認定遺伝カウンセラーによるカウンセリングでは、以下のような内容が扱われます。
- ➤遺伝形式と再発リスクの説明:KBG症候群の約3分の2は新生突然変異によるものですが、罹患した親から子へ伝わる場合の確率は理論上50%です。次子の妊娠を希望される場合、両親の遺伝学的検査と生殖細胞モザイクの可能性を含めた評価が必要です。
- ➤予後情報の率直な共有:表現型の幅が広く、軽症の方では知的能力が概ね保たれ、自動車の運転や買い物などの自立した生活スキルを獲得する成人例もあります。一方で、てんかんが制御不良の場合は家族や支援施設のサポートが生涯にわたって必要となります。
- ➤出生前診断の選択肢の提示:既知の変異が家系内にある場合、絨毛検査・羊水検査による胎児の確定診断が技術的に可能です。ただし「検査を受けることが常に正解」ではなく、ご夫婦の価値観に基づいた選択であることが大前提です。
- ➤心理的・社会的サポート:希少疾患であるため、診断直後のご家族は情報不足と孤立感に直面しがちです。患者支援団体「KBG Syndrome Association」「KBG Foundation」など、国際的なピアサポートの情報も合わせてご紹介しています。
なお、ミネルバクリニックのNIPT受検者の方々には互助会制度(8,000円)が適用され、陽性となった場合の羊水検査費用が全額補助されます。ANKRD11はインペリアルプランの検査対象遺伝子に含まれており、出生前の段階で評価することが可能な体制が整っています。
8. よくある誤解
誤解①「歯が大きいだけの病気」
巨歯症は最も特徴的な所見ですが、KBG症候群は生涯にわたって多臓器に影響する全身性の疾患です。新生児期の哺乳障害から成人期の大動脈拡張・強迫性障害まで、年齢ごとに異なる医学的課題が現れます。
誤解②「両親が健康なら遺伝病ではない」
KBG症候群の約3分の2は新生突然変異(de novo)によって発症します。両親に変異がなくても、お子さんに新しく生じた変異で発症することがあり、「両親が健康だから遺伝病ではない」という思い込みが診断を遅らせる原因になります。
誤解③「乳児期に診断がつかなければ違う」
KBG症候群は年齢依存的に表現型が進化する疾患です。巨歯症は永久歯が生え変わる7〜8歳頃まで確認できず、特徴的顔貌も成長とともに明瞭になります。乳児期に診断がつかなかった場合でも、学童期に再評価する意義は大きいです。
誤解④「強迫的なこだわりはIQと関係する」
KBG症候群でみられる強迫的特性は認知機能レベルとは独立して現れます。IQが保たれているお子さんでも、強い儀式的行動や強迫症状が日常生活を大きく妨げることがあります。行動療法や薬物療法を含む専門的な精神医学的評価が大切です。
9. 臨床遺伝専門医からのメッセージ
よくある質問(FAQ)
🏥 希少疾患の診断・遺伝カウンセリングについて
KBG症候群をはじめとする希少遺伝性疾患に関するご相談は、
臨床遺伝専門医が在籍するミネルバクリニックにお気軽にご相談ください。
関連記事
参考文献
- [1] Morel Swols D, Foster J 2nd, Tekin M. KBG Syndrome. GeneReviews®. University of Washington, Seattle. [NCBI Bookshelf NBK487886]
- [2] OMIM #148050. KBG Syndrome. Johns Hopkins University. [OMIM 148050]
- [3] Orphanet. KBG syndrome. ORPHA:2332. [Orphanet ORPHA:2332]
- [4] Liu B, Li Y, et al. ANKRD11 binding to cohesin suggests a connection between KBG syndrome and Cornelia de Lange syndrome. PNAS. 2025. [PNAS]
- [5] Low K, et al. Clinical and genetic aspects of KBG syndrome. Am J Med Genet A. 2016;170(11):2835-2846. [PubMed 27667800]
- [6] Walker M, et al. Life Beyond Childhood: Insight Into the Lived Experience of 91 Adults With KBG Syndrome. Am J Med Genet A. 2025. [PMC12066804]
- [7] Goldenberg A, et al. Natural history of KBG syndrome in a large European cohort. Hum Mol Genet. 2022;31(24):4131-4142. [Oxford Academic]
- [8] Guerin A, et al. Obsessive Compulsive Symptoms and Psychopathological Profile in Children and Adolescents with KBG Syndrome. Brain Sci. 2019. [PMC6895923]
- [9] ClinicalTrials.gov. Methylphenidate in KBG Syndrome: N-of-1 Series. NCT06465641. [ClinicalTrials.gov NCT06465641]
- [10] Isasi R, et al. KBG syndrome complicated with chylothorax in a newborn: a case report and literature review. Front Pediatr. 2025. [PMC12602497]
- [11] Isakov O, et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease. J Am Coll Cardiol. 2022. [ACC]
- [12] MedlinePlus Genetics. KBG syndrome. NIH National Library of Medicine. [MedlinePlus]






