目次

📍 クイックナビゲーション
12p重複症候群(トリソミー12p)は、第12染色体短腕(12p)の遺伝物質が過剰に存在することで起こる、出生5万人に1人とされる極めて稀少な染色体異常症候群です。1973年にUchidaとLinによって最初の症例が報告されて以来、半世紀にわたる細胞遺伝学・分子遺伝学の進歩によって病態の解明が大きく進みました。
中核となる症状は、特徴的な顔貌(丸い顔・突出した前頭部・低い鼻梁など)、重度の発達遅滞・知的障害、筋緊張の異常、難治性のてんかんです。一方で、重要な点として、過剰になっている遺伝物質の大きさ・モザイクの有無・含まれる原因遺伝子(GRIN2B、SCN8A、CHD4、ING4など)によって、症状の重さは患者さんごとに大きく異なります。
本記事では、12p重複症候群の発生メカニズム、原因遺伝子と症状の関係、Pallister-Killian症候群(テトラソミー12p)との重要な鑑別ポイント、染色体マイクロアレイ検査(CMA)による診断、そしてセノバメート・ラディプロディルなどの精密医療の最前線まで、臨床遺伝専門医の視点で網羅的にやさしく解説します。
1. 12p重複症候群(トリソミー12p)とは|疾患の基本情報
12p重複症候群は、第12染色体短腕(pアーム)の遺伝物質が通常の2コピーではなく3コピー(トリソミー)になることで発症する希少染色体異常です。「Duplication 12p」「Trisomy 12p」とも呼ばれます。出生5万人に1人と推定され、特徴的な頭蓋顔面部の異形性、重度の精神運動発達遅滞、軽度〜最重度の知的障害、筋緊張の異常、高頻度のてんかんを中核症状とします。
・トリソミー:染色体が通常2本のところ3本ある状態(全体または一部)。
・重複(duplication):染色体の一部分が余分にコピーされた状態。
12p重複症候群では、12番染色体の短腕の全部または一部が3コピーになります。含まれる遺伝子が「多すぎる」ことで、それぞれの遺伝子の働きが過剰になり、症状が出ます。これを「ドーズ効果(gene-dosage effect)」と呼びます。
1.1 疾患の概要
| 項目 | 内容 |
|---|---|
| 疾患名(日本語) | 12p重複症候群/トリソミー12p |
| 英語表記 | Duplication 12p syndrome / Trisomy 12p |
| 原因 | 第12染色体短腕(12p)の重複(部分的または全体) |
| 頻度 | 出生約5万人に1人(推定) |
| 遺伝形式 | 大半が新生突然変異(de novo)。一部は親の均衡型転座から不均衡型として遺伝 |
| 主な責任遺伝子(候補) | GRIN2B、SCN8A、CHD4、ING4、MFAP5、SOX5、ELKS/ERC1 など |
| 初の報告 | 1973年、Uchida & Lin による報告 |
1.2 Allenらによる5つのカテゴリー分類
1996年にAllenらによって、12p重複症候群は5つの主要なカテゴリーに分類されました。この分類は、患者さんの臨床的重症度や予後を予測する上で、現在も重要な枠組みです。
| カテゴリー | 細胞遺伝学的特徴 | 臨床的傾向 |
|---|---|---|
| カテゴリーⅠ | 12p11より遠位の「純粋な」部分トリソミー12p(他染色体異常なし) | 他のカテゴリーより予後良好、先天性奇形が少ない |
| カテゴリーⅡ | 正常細胞株と12pトリソミー細胞株が混在するモザイク型 | モザイク率に依存。完全トリソミーより予後が優れる傾向 |
| カテゴリーⅢ | 純粋な12pトリソミー+端部着糸型染色体(13・14・15・21・22番)の短腕異常 | 基本的に12pトリソミーの表現型が支配的 |
| カテゴリーⅣ | 完全12pトリソミー+非端部着糸型染色体のモノソミー/トリソミー(複合異常) | 胎児期致死や重篤な多発奇形のリスクが高い |
| カテゴリーⅤ | 完全12pトリソミー+同じ12番染色体長腕(12q)の異常 | 多指症などの骨格異常の追加リスク |
1.3 発生メカニズム|なぜ12pが重複するのか
12p重複の発生には、大きく分けて2つのメカニズムが知られています。
- 新生突然変異(de novo):大半の症例。両親に異常はなく、減数分裂や受精後の細胞分裂で偶発的に発生。非対立遺伝子相同組換え(NAHR)などの不均等な交差が原因と考えられています。
- 親の均衡型転座から派生:親が12番染色体を含む均衡型転座保因者の場合、減数分裂時の不分離により、不均衡型(12p重複+転座相手の欠失)として子に受け継がれます。この場合、他染色体異常を伴うため表現型が重篤になる傾向があります。
2. 12p重複症候群の主な症状|多系統への影響
12p重複症候群は、頭蓋顔面・中枢神経・骨格・歯科・感覚器など多系統に影響を及ぼします。一方で、心臓・腎臓・消化器など主要な内臓器官の重篤な奇形は比較的まれであり、これが本症候群の生命予後を全体として良好に保つ要因となっています。
2.1 主要症状の出現頻度
📊 12p重複症候群における主要症状の出現頻度
※Segelら2006・De Gregoriら2005・Liangら2006などの症例集積コホートに基づく概算値
2.2 中枢神経・発達への影響
本症候群の全例で発達遅滞がみられ、知的障害の程度は軽度から最重度まで幅広く分布します。モザイク型や狭い範囲の重複では比較的予後が良好な一方、完全な12p重複の患者さんでは重度〜最重度の知的障害を伴うことが一般的です。
- 運動発達:首のすわり・歩行などの粗大運動が著しく遅れ、自力歩行の獲得が4歳以降となることもあります。
- 言語発達:運動スキルよりも言語・発話の遅れがさらに顕著。生涯にわたり有意味語の獲得が困難なお子さんもいます。一方、言語の「理解力」は「話す力」よりも保たれている傾向があります。
- 気質・行動:「友好的で社交的」というプロフィールを示す方がいる一方、半数近くで自傷行為・激しい癇癪・睡眠障害が報告されています。ASDやADHDの併存例もあります。
2.3 特徴的な顔貌(craniofacial dysmorphism)
ほぼすべての患者さんで、12pの先端部分(12p13.2より末端)の遺伝子過剰に起因する一貫した顔貌の特徴が見られます。年齢とともに、乳幼児期の丸みを帯びた顔立ちから、成人期にかけてより「粗な顔立ち(coarse facial features)」へと変化することが報告されています。
- 頭部・上顔部:丸い顔、ふっくらとした頬、突出した前頭部、平坦な顔立ち
- 眼・鼻:両眼解離(hypertelorism)、内眼角贅皮、広く平坦な鼻梁、短い鼻、前を向いた鼻孔
- 口・顎:長く深い人中、口角が下がった大きな口、小顎症、薄い上唇/外反した下唇
- 耳・頸部:耳介低位、対輪の形態異常、短い首
2.4 てんかんと脳波(EEG)の特徴
てんかんは加齢に伴って発症率が上昇し、7歳以降の患者さんの多くが何らかの発作性疾患を有するようになります。乳児期から発症する早期発症例も存在します。発作型は欠神発作、ミオクロニー発作、全般性強直間代発作など多岐にわたります。
特徴的な脳波所見として、「3Hzの全般性棘徐波複合(3-Hz generalized spike and wave discharges)」が報告された症例があり、ミオクロニー発作を伴う症候性全般てんかんとして観察されることがあります。後述するように、GRIN2B・SCN8Aの過剰発現が関与する症例では、既存の抗てんかん薬に対する反応性が乏しい発達性およびてんかん性脳症(DEE)を呈するリスクが高くなります。
2.5 歯科・感覚器・外科的合併症
小児期以降に直面する深刻な課題のひとつが歯科的問題です。上顎・下顎のサイズ不一致による不正咬合、歯の萌出遅延、円錐歯・欠歯、エナメル質形成不全に加え、極めて高頻度で重度の齲蝕(虫歯)が進行します。障害児歯科による幼少期からの予防的介入が極めて重要です。
- 感覚器:反復性中耳炎から進行する滲出性中耳炎(glue ear)による伝音性難聴、兎眼による角膜乾燥
- 外科的:臍ヘルニア、男児の停留精巣、稀に十二指腸閉鎖などの消化管奇形
- 新生児期:哺乳障害、低血糖、稀に一過性糖尿病、低体温・呼吸不整・チアノーゼ

3. 原因遺伝子と分子メカニズム|なぜ症状が起こるのか
12p重複症候群の多彩な臨床像は、重複領域に存在する複数の遺伝子の「過剰発現」(ドーズ効果)に直接由来します。近年の高解像度マイクロアレイ解析により、12p内のどの領域がどの症状と関連するか、責任遺伝子のマッピングが急速に進展しました。
私たちの遺伝子は通常、父と母から1コピーずつ、計2コピーを持っています。重複によって遺伝子が3コピー(またはそれ以上)になると、遺伝子の働きが過剰になり、正常な発生・機能のバランスが崩れます。これがドーズ効果です。一方、片方のコピーが失われて1コピーになる「ハプロ不全」と並んで、遺伝子量の異常が引き起こす主要な病態メカニズムです。
3.1 主な責任遺伝子と役割
| 遺伝子 | 位置 | 主な役割と関連症状 |
|---|---|---|
| GRIN2B | 12p13.1 | NMDA受容体GluN2Bサブユニット。過剰発現でシナプスが過興奮し、発達性てんかん性脳症を引き起こす |
| SCN8A | 12p13.3 | 電位依存性Naチャネル(Nav1.6)。過剰発現で乳児期発症の難治性てんかん(DEE13) |
| CHD4 | 12p13.31 | クロマチン・リモデリング(NuRD複合体)。頭蓋顔面異常・脳発達異常に関与 |
| ING4・MFAP5 | 12p13.31 | 細胞増殖・アポトーシス・組織分化。「コア・フェノタイプ」の形成 |
| SOX5 | 12p12.1 | 転写因子(軟骨形成・神経堤分化)。重複で骨格異常・神経発達障害 |
| ELKS/ERC1 | 12p13.33 | シナプス活性帯因子。微細異常で小児期発症の言語失行(CAS) |
3.2 GRIN2B|NMDA受容体の過興奮による発達性てんかん性脳症
GRIN2B遺伝子は、脳の発生とシナプス可塑性に不可欠なNMDA受容体のGluN2Bサブユニットを作る設計図です。興味深いことに、この遺伝子は機能喪失(LoF)と機能獲得(GoF)で全く異なる病気を引き起こします。
- 機能喪失型(欠失や失活変異):知的障害・言語発達遅滞・多動・自閉症スペクトラム障害(ASD)が中心。発作は稀。
- 機能獲得型(12p重複によるコピー数増加):NMDA受容体の活動が過剰となり、シナプスの過興奮が神経毒性と異常同期を引き起こします。ウエスト症候群を含む重篤な発達性てんかん性脳症の直接的な原因となります。
3.3 SCN8A|難治性てんかんDEE13の原因
SCN8A遺伝子は、中枢神経系で重要な電位依存性ナトリウムチャネル(Nav1.6)を作ります。ニューロンが活動電位(電気信号)を発生させる際の主要なチャネルです。
GRIN2Bと同様、SCN8Aも機能獲得型と機能喪失型で表現型が二極化します。12p重複による過剰発現は強力な機能獲得をもたらし、通常生後数ヶ月以内に発症する極めて難治性の「発達性およびてんかん性脳症13型(DEE13)」を引き起こします。乳児痙攣・欠神発作・全般性強直間代発作など複数のタイプの発作が1日に数百回発生することもあり、既存の抗てんかん薬への抵抗性を示すことが多くなります。さらに、継続する発作による脳ダメージが、本来獲得していた運動・認知能力の「発達退行」を引き起こすという深刻な悪循環に陥ります。
3.4 12p13.31|頭蓋顔面異常の決定領域
特徴的な顔貌(丸い顔・突出した前頭部・低い鼻梁など)は、12pの遠位部、特に12p13.31領域の重複に強く依存すると考えられています。この領域にはING4・CHD4・MFAP5が含まれ、いずれも細胞増殖・組織分化・クロマチン制御に関わる中心的な遺伝子です。これらのコピー数が異常に増えることで、胎生期の頭蓋顔面構造形成や脳の構造的発達が阻害され、出生後の形態的・機能的異常へつながります。
3.5 12p13.33微細異常と「小児期発症の言語失行(CAS)」
染色体末端により近い12p13.33の微細な構造異常(微細欠失・微細重複)は、重度の言語表出の遅れや「小児期発症の言語失行(CAS)」と強く関連しています。CASは、発話に必要な筋肉に麻痺はないにもかかわらず、脳が発話の運動計画を正確にプログラムできないことで生じる障害です。この領域にはシナプス因子をコードするELKS/ERC1遺伝子が存在し、運動・構音ネットワークの正常な形成に重要な役割を果たします。
3.6 同じ12pでも「重複」と「欠失」では別の病気になる
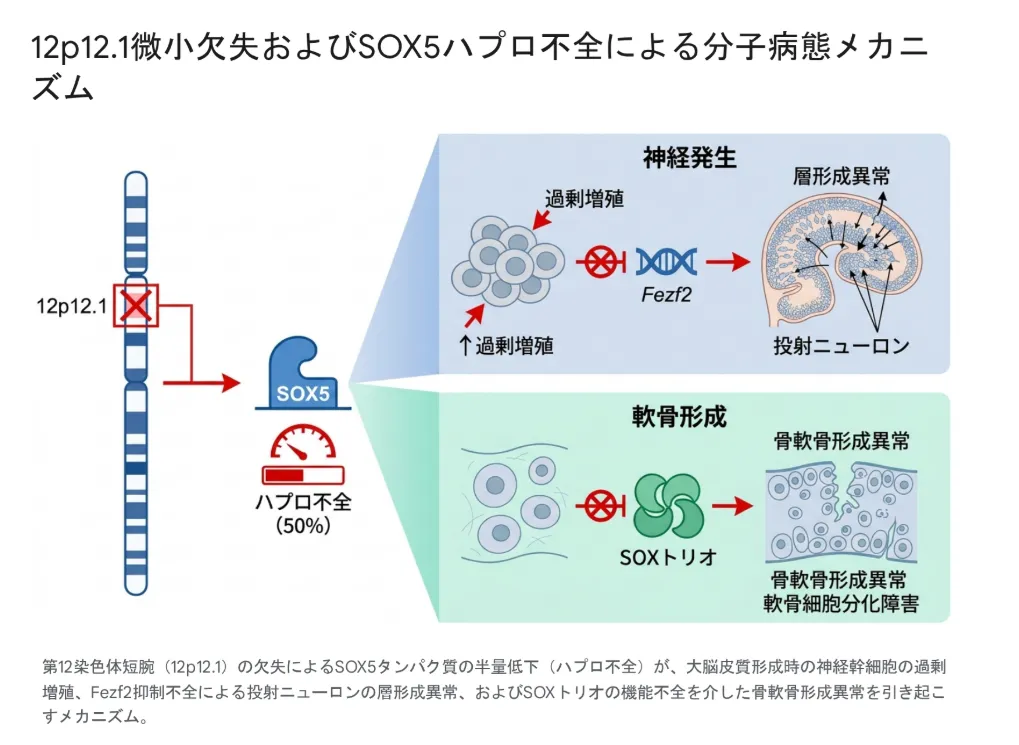
12pは、遺伝子が「過剰」になっても「不足」になっても、それぞれ違う症候群を引き起こします。たとえば12p12.1微小欠失症候群(Lamb-Shaffer症候群)は、SOX5遺伝子のハプロ不全が中心的な役割を担う独立した疾患です。同じ12p上で起きる遺伝子量の変化でも、「どの遺伝子が・どちらの方向に・どの程度」変化したかによって、病態は大きく変わります。
4. Pallister-Killian症候群との鑑別診断
12p重複症候群の臨床診断において、専門医が直面する最大の課題のひとつがPallister-Killian症候群(PKS)との鑑別です。両者は顔貌・発達遅滞の点で表面的に似通っていますが、遺伝学的基盤・モザイクの性質・伴う内臓奇形の重篤さが大きく異なる、明確に区別された疾患です。
4.1 遺伝学的基盤の決定的な違い
トリソミー12p vs テトラソミー12p(PKS)|臨床像の比較
4.2 PKSで「皮膚生検」が重要な理由
PKSの原因となる過剰な同腕染色体(i(12p))は、生後の末梢血リンパ球からは急速に失われる傾向があります。血液検査での検出率は0〜2%程度と極めて低く、通常の血液染色体検査では「正常」と誤診されるリスクが高いのです。一方、皮膚の線維芽細胞では50〜100%、羊水細胞では100%の高確率で保持されています。PKSを疑う場合は、皮膚生検や口腔粘膜スワブなど血液以外の組織を用いた解析が不可欠です。
このように、PKSはより広範で生命を直ちに脅かす多発奇形を伴うことが多い疾患である一方、純粋な12p重複症候群は、重度の知的障害を有しながらも主要な生命維持器官は正常であることが多く、全体的な生存率と身体的予後はPKSと比較して圧倒的に良好です。
5. 12p重複症候群の診断方法
本症候群の確定診断は、症状の多様性・重複サイズの微小性・PKSなどモザイク疾患との鑑別の必要性から、最新の分子遺伝学的手法を組み合わせた段階的アルゴリズムに沿って行われます。
5.1 出生後の確定診断|CMAがゴールドスタンダード
お子さんがすでに生まれており、ダウン症候群に似た顔貌だが21トリソミーではないお子さんや、原因不明の発達遅滞・知的障害・てんかんで医療機関を受診した場合、まず臨床評価で本症候群を疑い、血液検体を用いた染色体マイクロアレイ検査を行います。
CMA(chromosomal microarray analysis)は、従来のGバンド法では検出できない数kb〜数Mb単位の微小なコピー数異常を網羅的に検出する検査です。12p重複症候群では、例えば「arr 12p13.31-p11.1(6914072_34756209)×3」のように、約28Mbにわたる過剰部分を塩基対レベルの正確なブレイクポイントとともに同定できます。
5.2 検査方法ごとの違い
| 検査方法 | 特徴 | 12p重複への対応 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | 確定診断のゴールドスタンダード。微細CNVを高解像度で検出 | ◎ 第一選択 |
| Gバンド法(核型分析) | 解像度は約5〜10Mb | △ 大きな重複は検出可能だが、微小重複は見逃しやすい |
| サブテロメアMLPA法 | 特定染色体末端のコピー数を迅速にスクリーニング | ○ 初期スクリーニング用 |
| 頬粘膜スワブ/皮膚生検+CMA | 血液以外の組織を用いた評価 | ◎ PKS(テトラソミー)の鑑別に必須 |
| 全エクソームシーケンス(WES/CES) | SNVとCNVを同時解析 | △ 複雑症例の精査に有用 |
5.3 親への検査と再発リスク評価
確定診断後は、両親の血液検査(核型分析)が必須です。これにより、お子さんの異常が新生突然変異か親の均衡型転座由来かを判別し、将来の妊娠における再発リスクを正確に評価します。
| 状況 | 次のお子さんへの再発リスク |
|---|---|
| 両親とも異常なし(新生突然変異) | 原則として低い(1%未満)※生殖細胞モザイクの可能性は残る |
| 片親が12番染色体を含む均衡型転座保因者 | 転座の種類により高くなる。個別評価が必要 |
6. 治療と精密医療の最前線
現時点で12p重複症候群の根本的な遺伝子修復療法はありません。しかし、症状ごとの適切な対症療法・早期療育・継続的支援によって、生活の質を大きく改善することができます。さらに近年、難治性てんかんに対する「精密医療(Precision Medicine)」が急速に進歩しており、原因遺伝子のタイプに合わせた治療戦略が選択できるようになってきました。
6.1 多職種チームによる包括的管理
| ライフステージ | 主な対応 |
|---|---|
| 新生児期 | 哺乳支援、低血糖・体温管理、聴覚スクリーニング |
| 乳幼児期(〜5歳) | PT・OT・ST、AAC導入、滲出性中耳炎への鼓膜換気チューブ、停留精巣手術 |
| 学童期 | 特別支援教育、てんかん継続管理、障害児歯科による予防的介入 |
| 思春期・成人期 | 機能的生活スキル訓練、行動管理、レスパイトケア、家族支援 |
6.2 SCN8A過剰てんかんへの精密治療|ナトリウムチャネル阻害薬とセノバメート
SCN8A過剰発現による発達性てんかん性脳症(DEE13)は、一般的な抗てんかん薬には抵抗性を示しますが、病態がナトリウムチャネルの「過興奮(機能獲得)」に由来するため、これに合わせた特異的な薬剤選択が有効です。
- 高用量のナトリウムチャネル阻害薬:フェニトイン、オクスカルバゼピン、ラコサミドなどが選択的・特異的に効果を示すことが臨床データから確認されています。
- セノバメート(Cenobamate):成人焦点てんかん薬として開発されたセノバメートが、小児のSCN8A関連DEEに対しても極めて有効であることが初期の臨床報告から示されています。補助療法を受けた患者さんで発作頻度が劇的に減少し、一部では発作消失(seizure freedom)を達成。発作抑制に伴って覚醒度の向上など非発作性症状の改善も確認されています。
- 非薬物療法:薬物療法で十分なコントロールが得られない場合、迷走神経刺激療法(VNS)、反応性神経刺激(RNS)、ケトン食療法などが包括的に検討されます。
6.3 GRIN2B過剰への分子標的薬|ラディプロディルの臨床試験
NMDA受容体の異常亢進に対する次世代の分子標的薬の開発も進んでいます。ラディプロディル(Radiprodil)は、GluN2Bサブユニットの選択的ネガティブアロステリックモジュレーター(抑制剤)で、GRIN2Bの機能獲得型バリアントを有する患者さんを対象とした第1b/2a相試験(Honeycomb試験)で、優れた忍容性と発作頻度の実質的な減少、行動面での病態修飾効果を示す肯定的なトップラインデータが報告されました。
この知見をさらに確かめるため、現在は多施設共同・プラセボ対照二重盲検第3相試験(Beeline試験)がグローバルに進行中です。これらの治療法が確立されれば、12p重複患者さんの中枢神経症状への根本的な介入が可能になる可能性があります。
6.4 早期療育・長期予後・成人期への移行
重度の発達遅滞・運動発達遅滞に対しては、乳幼児期からの早期療育が長期予後に決定的な影響を与えます。理学療法(PT)・作業療法(OT)・言語聴覚療法(ST)に加え、発語獲得が困難な場合は絵カード交換式コミュニケーションシステム(PECS)、サイン言語、タブレット端末を用いた拡大代替コミュニケーション(AAC)を早期から導入することで、自己表出のフラストレーションを軽減し、行動障害の予防にもつながります。
成人期へのトランジションでは、学業の習得から「機能的生活スキル」の向上へとアプローチが変化します。多くの患者さんは食事・更衣・衛生管理に介助を必要としますが、健常な食物について学ぶ・身振りで症状を伝えるなど、実生活に即した訓練で自立性を高めることができます。家族(ケアギバー)へのレスパイトケアの充実、親亡き後を見据えたトランジション・プランニングへの法務・福祉的サポートも、社会システムとして重要です。
お子さんの発達や検査結果が気になっていませんか?
原因不明の発達遅滞・難治性てんかんには染色体マイクロアレイ検査が有効です。
臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
7. 出生前診断とミネルバクリニックのサポート体制
12p重複症候群は、NIPTのうち全染色体スクリーニング型のプラン(インペリアルプラン)でリスクを評価でき、羊水検査・絨毛検査でCMAを行うことで確定診断が可能です。ただし、表現型の幅が広く、出生前に見つけることが常にご家族の利益になるとは限らないため、検査前後の遺伝カウンセリングが不可欠です。
7.1 出生前検査の種類と検出能力
| 検査 | 位置づけ | 12p重複への対応 |
|---|---|---|
| NIPT(ターゲット型) | スクリーニング | △ 標準12微小欠失パネルには12p重複は含まれない |
| NIPT(全染色体5Mbスキャン) | スクリーニング | ○ 5Mb以上の12p重複をスクリーニング可能 |
| 絨毛検査+CMA | 確定診断 | ◎ 妊娠初期に確定診断可能 |
| 羊水検査+CMA | 確定診断 | ◎ 微小重複もブレイクポイントまで同定 |
7.2 ミネルバクリニックでのNIPTプラン
ミネルバクリニックでは、ご家族のニーズに合わせて複数のNIPTプランをご用意しています。ダイヤモンドプランはターゲット法による高精度検査で、当院が長年提供してきた特定12箇所の微小欠失(1p36、22q11.2、4p16など)を高い陽性的中率で検出しますが、12p重複はこの12箇所には含まれません。一方インペリアルプランはWGS法とターゲット法のハイブリッド構成で、5Mb以上の全染色体微小欠失・重複を広範囲にスクリーニングするため、12p重複領域もカバー対象となります。スクリーニング検査のため、陽性時は羊水検査・絨毛検査による確定診断が必要です。
7.3 出生前診断で12p重複が見つかった場合の対応
出生前に12p重複が指摘された場合、本症候群は表現型の幅が非常に広いため、胎児期の超音波所見だけで将来を正確に予測することは困難です。遺伝カウンセリングで重複範囲・関与する遺伝子・想定される症状の幅・予後の不確実性を中立的に説明し、両親の検査で新生突然変異か遺伝かを判定、詳細超音波で他染色体異常の合併・心奇形・脳の構造異常・骨格異常などを精査します。ご家族の不安や葛藤に寄り添い、決断を急がせない時間と環境を確保することが何より大切です。
⚖️ 倫理的なスタンス|検査は「常に利益」ではない
12p重複症候群のように表現型の幅が大きく、軽症から最重度まで分布する疾患では、出生前に見つけたことが必ずしもご家族の利益になるとは限りません。「特定の検査を勧める」「安心を保証する」「不安をあおる」表現は適切ではないと私たちは考えています。検査を受けるかどうか、結果をどう受け止めるかは、十分な情報を得たうえで、ご家族自身が決めるべき事柄です。
7.4 ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。12p重複症候群を含む染色体微小重複・微小欠失症候群について、出生前検査から結果説明、確定検査、その後のフォローまで一貫してサポートしています。
- 全染色体スクリーニング対応:インペリアルプランでは5Mb以上の全染色体微小欠失・重複を広くスクリーニング
- 確定検査も院内で実施:羊水検査・絨毛検査を院内で実施可能、転院の必要なし
- 臨床遺伝専門医が担当:臨床遺伝専門医が検査前後の遺伝カウンセリングを直接担当
- 互助会で費用面も安心:NIPT受検者全員に適用される互助会(8,000円)により、陽性時の羊水検査費用が全額補助
- 高精度の検査技術:当院のNIPTではCOATE法などの先進技術を採用
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
よくある質問(FAQ)
関連記事
参考文献
- GARD – Trisomy 12p [外部サイトへ]
- Unique – Duplications of 12p(rarechromo.org) [外部サイトへ]
- Arghir A et al. Pallister–Killian Syndrome versus Trisomy 12p—A Clinical Study of 5 New Cases and a Literature Review. Genes. 2021 [外部サイトへ]
- Non-mosaic partial duplication 12p in a patient with dysmorphic characteristics and developmental delay. PMC. [外部サイトへ]
- Segel R et al. The Natural History of Trisomy 12p. Am J Med Genet A. 2006 [外部サイトへ]
- Trisomy 12p syndrome: a chromosomal disorder associated with generalized 3-Hz spike and wave discharges. PubMed. [外部サイトへ]
- SCN8A-Related Epilepsy and/or Neurodevelopmental Disorders. GeneReviews. [外部サイトへ]
- Precision Medicine: SCN8A Encephalopathy Treated with Sodium Channel Blockers. PMC. [外部サイトへ]
- Cenobamate as add-on treatment for SCN8A developmental and epileptic encephalopathy. medRxiv 2024 [外部サイトへ]
- Opportunities for Precision Treatment of GRIN2A and GRIN2B Gain-of-Function Variants in Triheteromeric NMDA Receptors. PMC. [外部サイトへ]
- GRIN Therapeutics – Positive Topline Data from Honeycomb Trial of Radiprodil in GRIN-Related Neurodevelopmental Disorder. [外部サイトへ]
- 12p13.33 microdeletion including ELKS/ERC1, a new locus associated with childhood apraxia of speech. PMC. [外部サイトへ]
- Chromosome 12p Deletion Spanning the GRIN2B Gene Presenting With a Neurodevelopmental Phenotype. PMC. [外部サイトへ]
- Pallister-Killian Syndrome(CHOP). [外部サイトへ]






