目次


📍 クイックナビゲーション
12p12.1微小欠失症候群は、第12染色体短腕(12p12.1領域)の微小な欠失、または同領域内に位置するSOX5遺伝子のハプロ不全によって発症する希少な神経発達障害です。発見者の名前から「Lamb-Shaffer症候群(LAMSHF)」とも呼ばれ、SOX5ハプロ不全症候群と同義の疾患概念として扱われています。
中核症状は、著しい言語発達の遅れを伴う全般的な発達遅滞・中等度から重度の知的障害・自閉症様の行動特性です。特異的ながらも軽微な顔貌の特徴、骨格系の異形成、斜視などの眼科的合併症も高い頻度で見られます。世界で報告例は100〜113例規模の希少疾患ですが、染色体マイクロアレイ検査(CMA)や全エクソームシーケンス(WES)の臨床導入により、原因不明の発達遅滞として診断されていた症例から本症候群が次々と同定されています。
本記事では、最新の分子遺伝学的知見と長期追跡研究をもとに、12p12.1微小欠失症候群(Lamb-Shaffer症候群)の原因・症状・診断・治療・予後・遺伝カウンセリング・出生前診断の各論点を、臨床遺伝専門医の視点から網羅的に解説します。
1. 12p12.1微小欠失症候群とは|疾患の基本情報
12p12.1微小欠失症候群は、第12染色体短腕の12p12.1領域に生じる微小な染色体欠失を分子病態の基盤とする、極めて稀な先天性の神経発達障害および多発奇形症候群です。欠失領域に含まれる最も重要な責任遺伝子であるSOX5遺伝子のハプロ不全によって直接的に引き起こされることが明らかになっており、医学・遺伝学の文献では「Lamb-Shaffer症候群(LAMSHF)」「SOX5ハプロ不全症候群」と同義で扱われます。
中核症状は、言語表出の著しい遅滞を伴う全般的な発達遅滞、中等度から重度の知的障害、自閉スペクトラム症様の行動特性です。これに加えて、特異的な頭蓋顔面形態の異常、骨格系の異形成、眼科的疾患、稀ではあるがてんかん等の神経学的合併症を呈する多面的な症候群です。
私たちの遺伝子は、父と母から1コピーずつ、計2コピーずつ持っています。片方のコピーが欠失や機能喪失変異によって働かなくなり、残された1コピーだけでは生体が必要とする量のタンパク質を作れない状態を「ハプロ不全」と呼びます。SOX5のように発生過程で精緻な発現量制御を要する遺伝子は、用量感受性が高く、半量に減るだけで脳・骨格などの発生に大きな影響が出ます。
1.1 疾患の概要
| 項目 | 内容 |
|---|---|
| 疾患名 | 12p12.1微小欠失症候群/Lamb-Shaffer症候群/SOX5ハプロ不全症候群 |
| 英語表記 | 12p12.1 microdeletion syndrome/Lamb-Shaffer syndrome(LAMSHF) |
| 原因 | 第12染色体短腕(12p12.1)の微小欠失、またはSOX5遺伝子の機能喪失変異 |
| 頻度 | 100万人あたり1人未満(世界で約100〜113例の報告) |
| 遺伝形式 | 大半が新生突然変異(de novo)。常染色体顕性(優性)形式で稀に遺伝 |
| 主な責任遺伝子 | SOX5(中核)。隣接遺伝子としてPTHLH、CCDC91、PPFIBP1など |
| 国際分類 | OMIM:#616803、Orphanet:ORPHA 530983、MONDO:0014778 |
1.2 疾患認識の歴史
本症候群は、2012年にLambおよびShafferらによって独立した疾患概念として初めて定義されました。彼らは、マイクロアレイ染色体検査(aCGH)の普及に伴って見出された原因不明の知的障害を伴う患者群の中に、共通してSOX5遺伝子の欠失または変異を有する一群が存在することを発見しました。この発見以降、12p12.1領域の微小欠失とSOX5遺伝子内の点変異が同じ表現型スペクトラムを引き起こすことが確立され、疾患概念として整理されました。
近年は次世代シーケンシング(NGS)や全エクソームシーケンス(WES)が日常診療に導入されたことで、従来は原因不明の非特異的な発達遅滞と診断されていた患者さんの中から、SOX5の病的バリアントを有する症例が次々と同定されています。実際の有病率は報告例数を上回ると推測されており、今後さらに認知される疾患と考えられます。
1.3 名称の整理|「12p12.1微小欠失」と「Lamb-Shaffer症候群」は同じ疾患
医学文献やデータベースを参照する際、本症候群が複数の名称で呼ばれていることに戸惑われる方もいらっしゃいます。それぞれの名称は、疾患のどの側面に注目しているかの違いに過ぎず、本質的には同じ疾患を指しています。
- 12p12.1微小欠失症候群:染色体上の欠失位置に注目した呼称(病因の一つ)
- Lamb-Shaffer症候群(LAMSHF):2012年に疾患概念を確立したLambとShafferの名前を冠した正式な疾患名
- SOX5ハプロ不全症候群:最も重要な責任遺伝子であるSOX5の機能不全という分子病態に注目した呼称
12p12.1領域の微小欠失でSOX5が失われた場合も、SOX5遺伝子内の点変異や微小な挿入・欠失で機能が失われた場合も、最終的には同じ「SOX5ハプロ不全」という分子病態に収束し、共通の表現型を呈します。
2. 12p12.1微小欠失症候群の主な症状|多系統への影響
本症候群の臨床像は、中枢神経系を中心に、骨格系・眼科系・行動精神面など多系統に及びます。中でも、ほぼ全例で認められる言語発達遅滞と知的障害が中核症状となり、患者さんとご家族の生涯にわたる支援課題を形成します。一方で、てんかんや先天性心疾患などの致死的な合併症は他の重症染色体異常症と比べて頻度が高くないことも特徴的です。
2.1 主要症状の出現頻度(文献統合データに基づく推定)
📊 Lamb-Shaffer症候群における主要症状の出現頻度
※ 複数の臨床報告(PMC11285261、PMC9909913等)に基づく文献統合データの推定値。患者さんごとに大きな個人差があります。
2.2 認知・精神運動発達と言語機能
本症候群の最も高頻度かつ中核をなす臨床症状は、全般的な発達遅滞と知的障害です。運動発達のマイルストーン(首すわり・寝返り・お座りなど)は総じて遅延するものの、大多数の患児は2〜3歳の間に何らかの形で自立歩行を獲得すると報告されています。
一方で、言語発達の遅れは極めて深刻で、本症候群を決定づける特徴の一つです。初めて意味のある言葉を発するのは1歳半から4歳と非常に遅く、言葉を獲得しても2語文程度の限定的な表現にとどまるお子さんが多くいらっしゃいます。少数ながら生涯にわたり発語を獲得できず、マカトンサインや絵カード、タブレット型のAAC(拡大代替コミュニケーション)に依存するケースも存在します。理解言語(受容言語)の能力は、表出言語に比べて相対的に保たれる傾向があり、これが学齢期以降の特別支援教育を組み立てるうえで重要な手がかりとなります。
2.3 行動・精神医学的特徴
12p間質性欠失スペクトラムは、自閉スペクトラム症(ASD)や関連する発達障害の素因となる遺伝的背景として国際的に認識されています。患者さんはしばしば、社会的相互作用の難しさ、限定的・反復的な興味、視線が合いにくいといった自閉症様の行動特性を示します。
- 多動・注意の課題:ADHDに類似した多動性・短い注意力
- 常同行動:意味のない反復的な運動や発声を伴うことがある
- 低い欲求不満耐性:激しい癇癪(temper tantrums)や予測困難な攻撃性の爆発
- 感覚過敏:特定の音響や衣服の触感に対する強い反応
- 不安傾向:新しい環境や予定変更への適応の難しさ
2.4 頭蓋顔面形態の特徴|軽微なため見過ごされやすい
本症候群の患者さんには、軽度ながら共通する顔貌の特徴(dysmorphic features)が見られます。ダウン症候群などの古典的な染色体異常症と比較すると、特徴が必ずしも顕著ではなく、家族や医療者にも見過ごされる場合があります。この「軽微さ」こそが、本症候群が長らく原因不明の発達遅滞として分類されてきた一因でもあります。
- 頭部:前頭部の突出(frontal bossing)、両眼隔離(hypertelorism)
- 眼:眼裂の斜下(downslanting palpebral fissures)、内眼角贅皮(epicanthal folds)
- 鼻:鼻根部の平坦化、球状の鼻尖(bulbous nasal tip)
- 口・顎:薄い上唇または不規則に厚い口唇、小さな顎(micrognathia)
- 耳:耳輪の低形成を伴う低位耳・後方回転耳
これらの顔面所見は成長とともに「粗造化(coarsening)」していくというダイナミックな変化を示すことが、近年のイタリアにおける長期追跡コホート研究で明らかになっています。詳細は後述します。
2.5 筋骨格系と運動機能
乳児期初期から見られる全身の筋緊張低下(hypotonia)は、運動発達遅滞の主要な要因となります。筋力不足と協調運動の困難さにより、スプーンの使用・ボタンかけ・鉛筆の把持などの微細運動スキルの獲得に顕著な困難を伴います。
骨格系の異常はSOX5そのものに加えて、隣接するPTHLH・CCDC91などの遺伝子の機能不全が複合的に関与しているとされ、成長とともに顕在化します。
- 脊柱の弯曲異常:側弯症(scoliosis)・後弯症(kyphosis)は約30%に認められ、成長期に進行
- 椎体の形態異常:蝶形椎(butterfly vertebrae)など
- 胸郭の変形:漏斗胸や鳩胸(pectus carinatum)
- 四肢:手足の指の斜指症(clinodactyly)、関節の過可動性(hyperlaxity)
- 足部:足の外反変形が報告される症例も
2.6 眼科・摂食・てんかんなど他の合併症
- 眼科的異常:斜視(内斜視・外斜視)が約40%と高頻度。視神経低形成、皮質盲、屈折異常も
- 摂食障害:新生児期に筋緊張低下と口腔咽頭筋の協調不全による哺乳障害。経管栄養を要するケースも
- 消化器症状:慢性的かつ頑固な便秘が高頻度に見られる
- てんかん:約22%(コホートにより10〜25%程度)。発症した場合は欠神発作・全般強直間代発作など多様な発作型
3. 原因遺伝子SOX5|なぜ症状が起こるのか
12p12.1微小欠失症候群の症状は、SOX5遺伝子のハプロ不全を分子病態の中核としています。SOX5は神経幹細胞の制御や大脳皮質の形成、軟骨形成など、発生過程で不可欠な役割を担う「マスター転写因子」であり、その発現量が半分になっただけで広範な発生学的影響が現れます。
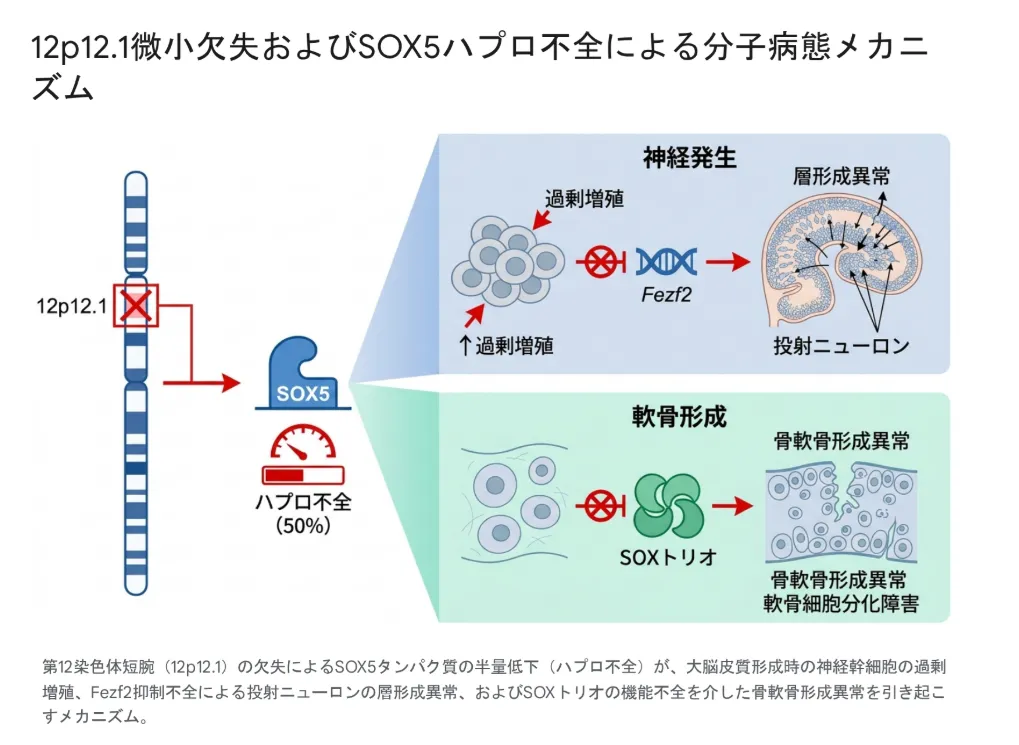
図:12p12.1領域の欠失によるSOX5タンパク質の半量低下(ハプロ不全)が、大脳皮質形成時の神経幹細胞の過剰増殖、Fezf2抑制不全による投射ニューロンの層形成異常、およびSOXトリオの機能不全を介した骨軟骨形成異常を引き起こすメカニズム。
3.1 SOX5とはどんな遺伝子か
SOX5はSOX(SRY-related HMG-box)転写因子ファミリーに属する遺伝子で、DNAに直接結合して他の遺伝子のオン・オフを切り替える「指揮者」のような働きをします。神経・骨格・心臓など多くの組織で、発生のタイミングを精密にコントロールする中心的な役割を担うため、量が変動すると影響が広範囲に及びます。
3.2 神経発生におけるSOX5の役割
SOX5は脳と神経系の発生において、時期・部位特異的な制御を行います。ハプロ不全による分子病態は主に次の二つの経路で説明されます。
① 神経幹細胞の「休眠」維持の破綻:生まれてから2週目あたりの時期に、SOX5は神経幹細胞のBMPシグナル経路を制御し、幹細胞を適切な「休眠状態」に保ちます。SOX5が半量になると、神経幹細胞が浅い休眠状態のまま過剰に増殖し、その反動で成体期の神経幹細胞プールが早期に枯渇します。結果として長期的な神経新生能力が損なわれます。
② 大脳皮質深層ニューロンの層形成異常:胎生期の大脳皮質形成において、SOX5は深層(第5層・第6層)に定着すべきニューロンに対してFezf2やBcl11bといった転写因子の発現を適切に抑制します。SOX5が欠損すると、これらの遺伝子の抑制に失敗し、深層ニューロンが分子特性の混在した異常な細胞へと分化し、皮質下への投射パターンに致命的な配線エラーを生じます。この大脳皮質の微細構造異常こそが、患者さんの重篤な知的障害・言語発達遅滞・自閉症様行動の器質的基盤と考えられています。
3.3 骨軟骨形成への影響|SOXトリオ
SOX5は神経系だけでなく、軟骨細胞の分化と細胞外基質の産生を司る主要な調節因子でもあります。同ファミリーのSOX6・SOX9とともに「SOXトリオ」と呼ばれる複合体を形成し、軟骨に特有の遺伝子(アグリカンなど)を相乗的に活性化します。
SOX5のハプロ不全はSOXトリオの機能を不全に陥らせ、長管骨や脊椎の形成に不可欠な「内軟骨性骨化」を阻害します。これが、患者さんに特徴的な側弯症・蝶形椎・胸郭異常の分子基盤です。
3.4 隣接遺伝子欠失症候群としての側面
12p12.1微小欠失症候群は、欠失サイズによってはSOX5以外の隣接遺伝子も同時に失われる「隣接遺伝子欠失症候群(contiguous gene deletion syndrome)」としての側面を持ちます。欠失範囲によって表現型の重症度や合併症のパターンが変動するため、欠失境界(ブレイクポイント)の正確な同定が予後評価に重要です。
| 遺伝子 | 主な役割 | 欠失時の表現型 |
|---|---|---|
| SOX5 | 神経幹細胞制御、大脳皮質層形成、SOXトリオの軟骨形成 | 知的障害、言語遅滞、自閉症様行動、骨格異常(中核的責任遺伝子) |
| PTHLH | 副甲状腺ホルモン関連タンパク質、内軟骨性骨化を強力に調節 | 短指症E2型(brachydactyly E2)、鳩胸、早発性骨端線閉鎖 |
| CCDC91 | ハプロ不全不耐応遺伝子(用量感受性が高い) | PTHLHと協調的に骨格異常の発症に関与 |
| PPFIBP1 | 神経発達関連 | 優性的な神経発達障害の増悪因子としての関与が示唆 |
3.5 遺伝形式と再発リスク
・常染色体顕性(優性):2022年に日本人類遺伝学会で「優性遺伝」が「顕性遺伝」、「劣性遺伝」が「潜性遺伝」へと用語変更されました。本症候群は、片方の染色体に欠失や変異があるだけで発症する常染色体顕性形式をとります。
・新生突然変異(de novo):両親には欠失・変異がなく、お子さんで新たに変異が発生したケースを指します。本症候群の大半はこの新生突然変異で生じます。
本症候群の大多数は新生突然変異によって生じるため、同じご両親から次のお子さんが本症候群を発症する再発リスクは1%未満とされています。しかし、一部の家系では遺伝例も報告されており、片親が軽度の表現型を伴うSOX5病的バリアントを持っていたり、生殖細胞モザイクの状態で保有しているケースが確認されています。重度の知的障害を有する兄弟例で、一見健康な父親がSOX5欠失の体細胞・生殖細胞系列モザイクであったというイタリアの報告は、「孤発性に見える神経発達障害」の背後にモザイク変異が潜む可能性を示す重要な事例です。
このため、お子さんの欠失が同定された場合は、両親のSOX5検査と高感度のモザイク検出が再発リスク評価に不可欠です。
4. 診断方法と鑑別診断|CMAとWESの両輪
12p12.1微小欠失症候群は、特異的な軽微な顔貌・重度言語遅滞・自閉症様行動などの臨床所見から疑うことはできますが、表現型が多岐にわたるため臨床所見のみでの確定診断は不可能です。確定診断には分子遺伝学的検査が必須となります。
4.1 出生後の確定診断|CMAとWESの組み合わせ
・CMA:数キロベース〜数メガベース単位の微小な染色体欠失・重複(コピー数変異:CNV)を網羅的に検出。従来のGバンド法では捉えられない微小な欠失を検出する第一線の検査です。
・WES:遺伝子のタンパク質コーディング領域全体の塩基配列レベルの異常(ナンセンス変異、ミスセンス変異、スプライシング異常など)を網羅的に解析。両親と本人を同時解析するTrio-WESにより、変異が新生突然変異か遺伝性かを即時に判定できます。
本症候群の診断戦略は、「まずCMAで12p12.1領域の欠失を探し、CMAで陰性ならWESでSOX5遺伝子内部の点変異を探す」という二段階アプローチが基本です。両親の血液で同じ変異の有無を確認し(新生突然変異か遺伝かを判定)、頭部MRI・脳波(EEG)・心エコー・眼科診察などで合併症を精査します。
4.2 検査方法ごとの違い
| 検査方法 | 特徴 | 12p12.1欠失/SOX5変異の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | 微細CNVを高解像度で検出 | ◎ 欠失型は確実に検出 |
| 全エクソームシーケンス(WES) | 遺伝子の塩基配列を網羅的に解析 | ◎ SOX5内部の点変異も検出 |
| Gバンド法(核型分析) | 解像度は約5〜10Mb | ✕ 検出困難(微小欠失は見逃される) |
| FISH法 | SOX5プローブで迅速確認 | △ 専用プローブで欠失型のみ可能 |
4.3 鑑別診断|似た症状を示す疾患群
本症候群は症状が多彩で軽微な顔貌異常を伴うため、初期評価では他の神経発達障害症候群との鑑別が必要となります。
- 原因不明の発達遅滞・知的障害:最も多い「鑑別前段階」。CMAとWESで本症候群が同定されるケースが多数
- 自閉スペクトラム症(ASD):行動特性が重なるが、本症候群では言語遅滞・軽微な顔貌・筋緊張低下が併存
- 他の微小欠失症候群:2q33欠失症候群(SATB2関連症候群)、17q21.31欠失症候群など、知的障害+言語遅滞を呈する疾患群との鑑別
- PTHLH関連の短指症E2型:12p12.1領域内のPTHLH単独欠失例では知的・神経発達は年齢相応で、骨格異常が前景に立つ
お子さんの発達や検査結果が気になっていませんか?
原因不明の発達遅滞・言語遅延・自閉症様行動には、染色体マイクロアレイ検査と全エクソームシーケンスが有効です。
臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
5. 治療と長期管理|早期療育と多職種連携
12p12.1微小欠失症候群そのものを根治する治療法(薬物療法・酵素補充療法・遺伝子治療)は現時点で確立されていません。管理の主眼は、各患者さんが持つ個別の症状に対する対症療法と、認知・運動能力のポテンシャルを最大限に引き出すための早期からの包括的・多職種介入に置かれます。
5.1 早期療育(PT・OT・ST)
神経発達の可塑性が高い乳幼児期からの介入が、長期的な発達と生活の質に大きく影響します。発達小児科医・小児神経科医・臨床心理士の主導のもと、診断確定後できるだけ早期から療育プログラムを開始することが専門家から強く推奨されています。
- 言語聴覚療法(ST):構音訓練、発語促進、早期からのAAC(拡大代替コミュニケーション)導入
- 理学療法(PT):筋緊張低下への対応、粗大運動の獲得支援、側弯症の進行予防
- 作業療法(OT):微細運動・食事・着替えなど日常生活動作(ADL)の習得
- 個別教育計画(IEP):学齢期は個々の認知プロファイルを評価し、視覚的・段階的に教示する学習アプローチを策定
5.2 ライフステージ別の管理
| ライフステージ | 主な対応 |
|---|---|
| 新生児期・乳児期 | 哺乳障害への栄養支援、筋緊張低下のフォロー、眼科スクリーニング |
| 幼児期(〜5歳) | 早期療育(PT・OT・ST)、AAC導入、便秘管理、てんかんがあれば抗てんかん薬 |
| 学童期(6〜12歳) | 特別支援教育(IEP)、側弯症の整形外科的フォロー、ABA等の行動療法 |
| 思春期・成人期 | 移行期医療、攻撃性・癇癪への薬理学的介入の検討、生活自立支援、就労支援 |
5.3 行動面の管理
ADHD様の多動性・衝動性、自閉症様の社会的相互作用の難しさ、激しい癇癪などに対しては、応用行動分析(ABA)などの行動療法をベースに、必要に応じて非定型抗精神病薬や中枢神経刺激薬による薬理学的介入が検討されます。後述する通り、成人期に向けて行動面の課題が顕在化していくことが報告されているため、早期からの介入と継続的な評価が重要です。
5.4 整形外科・てんかん・消化器の管理
- 整形外科:側弯症は成長期に急速に進行するリスクがあり、定期的なX線評価と装具療法、重症例では脊椎固定術を検討
- てんかん:発作型に応じた抗てんかん薬(AEDs)の選択、定期的な脳波モニタリング
- 消化器:慢性便秘に対する食物繊維増量・浸透圧性下剤による排便コントロール
- 眼科:斜視矯正術、屈折異常への眼鏡処方、定期的な視機能評価
5.5 長期予後と成人期の表現型の変化
本症候群が疾患概念として確立してから日が浅いため、成人に達した患者さんの長期的な自然歴は限定的です。現時点での医学的コンセンサスとしては、心臓の重篤な構造異常など生命予後を直接脅かす合併症は稀で、疾患そのものが寿命に直接的な悪影響を及ぼす証拠はないとされています。
一方で、近年のイタリアでの長期追跡研究(Innellaらの報告)により、成人期に向けて顔貌が「粗造化」し、行動面の課題が増悪するというダイナミックな表現型の変化が記録されています。
| 評価ドメイン | 幼児・学童期 | 成人期(30歳代) |
|---|---|---|
| 顔貌 | 薄い鼻と唇、逆三角形の小さな顔立ち | 顔貌全体が粗造化、高く広い前頭部、歯肉増殖、下顎の突出 |
| 頭囲 | 出生時は概ね正常 | 相対的な小頭症が顕在化 |
| 行動・精神 | 多動、衝動、頻繁な癇癪 | 攻撃性の爆発が増悪、専門施設入所例も |
| 言語・認知 | 重度言語遅滞、限定的語彙 | 表出言語の貧弱さが固定化、知的障害が持続 |
この経時的な変化は、SOX5が単なる胎生期初期の形態形成のトリガーではなく、生涯にわたり脳神経回路の維持・結合組織のモデリング・精神的ホメオスタシスに継続的に関与している可能性を示唆しています。生涯にわたる包括的ケアの体制づくりが必要です。
6. 遺伝カウンセリングと再発リスク
12p12.1微小欠失症候群は表現型の幅が広く、同じ変異でも症状の出方や重症度が異なることが知られています。遺伝カウンセリングを通じて、ご家族が病気を正確に理解し、中立的な情報のもとに納得のいく決断ができるよう情報提供することが、医師の重要な役割となります。
6.1 カウンセリングで伝えるべきポイント
- 欠失範囲と表現型の関係:含まれる遺伝子(PTHLHを含むかなど)によって骨格症状の重さが変わる
- 表現型の多様性:同じSOX5変異でも症状の出方は予測が難しい
- 成人期の見通し:顔貌の粗造化や行動面の課題の増悪を見据えた生涯支援計画
- 両親の検査:新生突然変異か遺伝かを判定。生殖細胞モザイクの可能性も考慮
- 支援体制:多職種チーム、療育、社会福祉制度、海外の患者支援団体(Unique・Simons Searchlight)
6.2 再発リスク
| 状況 | 次のお子さんへの再発リスク |
|---|---|
| 両親とも欠失なし(新生突然変異) | 原則として低い(1%未満)※生殖細胞モザイクの可能性は残る |
| 片親が保因者 | 理論的に50%(表現型の幅が大きく、症状の出方は予測困難) |
| 片親が生殖細胞モザイク | 「孤発例」と思われた家系で兄弟例が発症するケースあり。高感度検査が必要 |
7. 出生前診断とミネルバクリニックのサポート体制
12p12.1微小欠失症候群は、NIPTのうち全染色体スクリーニング型のプラン(インペリアルプラン)でリスクを評価でき、羊水検査・絨毛検査でCMAを行うことで確定診断ができます。ただし、本症候群は表現型の幅が広く、出生前に見つけることが常にご家族の利益になるとは限らないため、検査前後の遺伝カウンセリングが不可欠です。
7.1 出生前検査の種類と検出能力
| 検査 | 位置づけ | 12p12.1欠失への対応 |
|---|---|---|
| NIPT(ターゲット型) | スクリーニング検査 | 対象外(特定12微小欠失のみが対象のプランでは12p12.1は含まれない) |
| NIPT(インペリアルプラン) | スクリーニング検査 | ○ スクリーニング可能(5Mb以上の全染色体微小欠失をWGSで網羅、12p12.1領域もカバー対象) |
| 絨毛検査+CMA | 確定診断 | ◎ 妊娠初期に確定診断可能 |
| 羊水検査+CMA | 確定診断 | ◎ 微小欠失も確定診断 |
7.2 ミネルバクリニックでのNIPTプランと12p12.1欠失
ミネルバクリニックでは、ご家族のニーズに応じて複数のNIPTプランをご用意しています。ダイヤモンドプランはターゲット法による高精度検査で、特定12箇所の微小欠失(1p36、22q11.2など)を高い陽性的中率で検出しますが、12p12.1はこの12箇所には含まれません。一方インペリアルプランはWGS法とターゲット法のハイブリッドで、5Mb以上の全染色体微小欠失・重複を広範囲にスクリーニングするため、12p12.1領域もカバー対象となります。
ただしNIPTはあくまでスクリーニング検査のため、陽性となった場合は羊水検査・絨毛検査とCMAによる確定診断が必要です。
7.3 出生前診断で見つかった場合の対応
出生前に12p12.1欠失が見つかった場合、本症候群は表現型の幅が非常に広く、また顔貌異常は軽微であるため胎児期の超音波所見だけで予後を予測することは困難です。遺伝カウンセリングで欠失範囲・関与する遺伝子・表現型の幅・予後の不確実性を中立的に説明し、両親の検査で新生突然変異か遺伝かを判定、詳細超音波で骨格・脳・眼などを精査します。ご家族の不安や葛藤に寄り添い、決断を急がせない時間と環境を確保することが何より重要です。
⚖️ 倫理的なスタンス|検査は「常に利益」ではない
12p12.1微小欠失症候群のように表現型の幅が大きい疾患では、出生前に見つけたことが必ずしもご家族の利益になるとは限りません。「特定の検査を勧める」「安心を保証する」「不安をあおる」ような表現は適切ではないと私たちは考えています。検査を受けるかどうか、結果をどう受け止めるかは、十分な情報を得たうえで、ご家族自身が決めるべき事柄です。
7.4 ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。12p12.1微小欠失症候群を含む染色体微小欠失症候群について、出生前検査から結果説明、確定検査、その後のフォローまで一貫してサポートいたします。
- 全染色体スクリーニング対応:インペリアルプランでは5Mb以上の全染色体微小欠失・重複を広くスクリーニング、12p12.1領域もカバー対象
- 高精度検査も選択可:COATE法を採用したターゲット型検査で、対象疾患は高い陽性的中率を実現
- 確定検査も院内で実施:羊水検査・絨毛検査も院内で実施可能、転院の必要なし
- 臨床遺伝専門医が担当:臨床遺伝専門医が検査前後の遺伝カウンセリングを直接担当
- 互助会で費用面も安心:NIPT受検者全員に適用される互助会(8,000円)により、陽性時の羊水検査費用が全額補助
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
よくある質問(FAQ)
関連記事
参考文献
- OMIM #616803 – Lamb-Shaffer Syndrome(LAMSHF) [外部サイトへ]
- Orphanet – Lamb-Shaffer syndrome (ORPHA:530983) [外部サイトへ]
- NCBI MedGen – 12p12.1 microdeletion syndrome (C4755260) [外部サイトへ]
- Lamb AN, Rosenfeld JA et al. Haploinsufficiency of SOX5 at 12p12.1 is associated with developmental delays with prominent language delay, behavior problems, and mild dysmorphic features. Hum Mutat. 2012 [外部サイトへ]
- Zhang B et al. Clinical cases series and pathogenesis of Lamb-Shaffer syndrome. 2024 [外部サイトへ]
- Clinical characterization of Lamb-Shaffer syndrome: a case report and literature review. 2023 [外部サイトへ]
- Innella G et al. Clinical spectrum and follow-up in six individuals with Lamb-Shaffer syndrome (SOX5). Am J Med Genet A. 2021 [外部サイトへ]
- Interstitial 12p Deletion Syndrome: Revised Minimal Critical Region and Review of the Literature. Genes (MDPI). 2026 [外部サイトへ]
- Lai T et al. SOX5 controls the establishment of quiescence in neural stem cells during postnatal development. PMC. 2024 [外部サイトへ]
- Kwan KY et al. SOX5 postmitotically regulates migration, postmigratory differentiation, and projections of subplate and deep-layer neocortical neurons. PNAS. 2008 [外部サイトへ]
- Cleveland Clinic – Lamb-Shaffer Syndrome: Causes, Symptoms & Treatment [外部サイトへ]
- Simons Searchlight – SOX5-related syndrome (Gene Guide) [外部サイトへ]
- Unique – Rare Chromosome Disorder Support Group: SOX5 syndrome (Lamb-Shaffer Syndrome) factsheet [外部サイトへ]
- Lamb-Shaffer Research Foundation [外部サイトへ]






