目次


📍 クイックナビゲーション
8p重複症候群は、第8番染色体短腕(8p)に余分な遺伝物質が存在することで発症する、極めて稀少な染色体構造異常の総称です。重複の範囲や形態によって表現型は大きく異なりますが、中等度から重度の発達遅滞・知的障害・特徴的な顔貌・脳梁の低形成・先天性心疾患・てんかんなどを主症状とし、多系統に影響が及ぶことが知られています。
従来のGバンド染色体検査では見逃されてきた症例が、染色体マイクロアレイ検査(CMA)の臨床導入に伴って次々と診断されるようになり、独立した疾患群として確立されてきました。8p23.1領域に存在する嗅覚受容体遺伝子クラスターを介した非対立遺伝子間相同組換え(NAHR)が主要な発症メカニズムであり、ヨーロッパ系人口の約26%が保有するパラセントリック逆位多型が、次世代における重篤な再編成のリスク因子となっていることも明らかになっています。
本記事では、2025年に発表されたSantucciらによる最新の臨床管理ガイドラインを含め、8p重複症候群の原因・症状・診断・治療・予後・遺伝カウンセリング・出生前診断について、臨床遺伝専門医の視点から網羅的に解説します。
1. 8p重複症候群とは|疾患の基本情報
8p重複症候群は、第8番染色体の短腕(p腕)に余分な遺伝物質が存在することで発症する稀少な染色体構造異常の総称です。広義の「8p重複」には、短腕全体の重複(8pトリソミー)、特定領域の微小重複(8p23.1重複症候群)、重複と末端欠失が複合した8p逆位重複欠失症候群(invdupdel(8p))、モザイク型、環状染色体型まで含まれ、関与する染色体セグメントのサイズ・位置・包含される遺伝子群によって表現型が大きく変わるのが最大の特徴です。
染色体には「セントロメア」と呼ばれるくびれがあり、これを境に短い腕を「p腕(short=petitに由来)」、長い腕を「q腕」と呼びます。第8番染色体は約1億4,600万塩基対の長さを持ち、短腕(8p)は約44メガベース(Mb)の長さに484の遺伝子をコードしています。8p重複症候群ではこのp腕の一部または全体に余分なコピーが生じ、その範囲内の遺伝子量が増えることで多臓器に影響が現れます。
1.1 疾患の概要
| 項目 | 内容 |
|---|---|
| 疾患群名 | 8p重複症候群(第8番染色体短腕重複症候群) |
| 英語表記 | Chromosome 8p duplication syndrome / 8p23.1 microduplication syndrome / Inverted duplication-deletion of 8p(invdupdel(8p)) |
| 原因 | 第8番染色体短腕の余剰な遺伝物質(重複) |
| 頻度 | invdupdel(8p):約1/10,000〜1/30,000出生、8p23.1微小重複症候群:約1/58,000出生 |
| 遺伝形式 | 大半が新生突然変異(de novo)。親が無症候性の逆位多型を保有する場合は次世代でリスク上昇 |
| 主な責任遺伝子・領域 | 8p23.1領域(GATA4、SOX7、TNKS1、MIR124-1など32遺伝子) |
| 国際分類 | Orphanet:96092(invdupdel(8p))/251076(8p23.1重複症候群)、GARD:5361/10304 |
1.2 主要な構造異常の類型
「8p重複」と一口に言っても、実際には複数のサブタイプが存在し、それぞれ重症度や合併症のパターンが異なります。臨床現場でよく遭遇する4つの類型を整理しておきましょう。
| 類型 | 特徴 | 臨床像の傾向 |
|---|---|---|
| 8p逆位重複欠失症候群(invdupdel(8p)) | 遠位の末端欠失+中間の正常領域+近位の逆位重複が複合 | 最も重症。脳梁低形成・欠損を約80%に伴う |
| 8p23.1微小重複症候群 | 8p23.1領域(約3.68Mb・32遺伝子を含む)の純粋な重複 | 認知機能障害・自閉傾向が中心。脳梁欠損などはinvdupdel(8p)より少ない |
| モザイク型8pトリソミー | 8p重複細胞と正常細胞が個体内に混在 | 異常細胞の割合・分布で症状の幅が大きい。完全型より軽症のことが多い |
| 8p環状染色体 | 両端が結合してリング状になった構造異常 | 末端欠失+細胞分裂時の不安定性によりモザイク化、重篤な成長障害を伴うことあり |
1.3 疾患認識の歴史と現状
8p重複と欠失の複合病態は、1976年にWeleberらによって医学文献に初めて記載されました。その後、染色体マイクロアレイ検査(CMA)の臨床導入によって、Gバンド染色体検査では見逃されていた軽症例や非典型例が次々と診断されるようになり、患者数は大きく増加しています。
歴史的には純粋なinvdupdel(8p)の報告例は約60例とされてきましたが、現在では関連患者コミュニティやレジストリにおいて世界で350名以上の患者が確認されており、実際の有病率は過去の推定を上回る可能性が示唆されています。2018年に設立されたProject 8p Foundationは、患者支援と研究推進を一体化した国際的なハブとして機能しており、診療ガイドラインの整備・iPSCを用いた疾患モデルの開発・染色体工学を用いた将来的な治療戦略まで、幅広い取り組みを進めています。
2. 8p重複症候群の主な症状|多系統への影響
8p重複症候群は単一臓器の病気ではなく、中枢神経系・頭蓋顔面・心血管系・骨格系・消化器系・内分泌系など多系統に影響します。中でも中等度〜重度の発達遅滞・知的障害はほぼ全例に認められる中核症状であり、invdupdel(8p)の場合は脳梁の低形成・欠損と先天性心疾患が長期予後を大きく左右します。
2.1 主要症状の出現頻度
📊 8p重複症候群における主要症状の出現頻度
※invdupdel(8p)を中心とした臨床コホート・症例集積からの推定値。重複範囲・サブタイプにより大きく変動します。
2.2 中枢神経・神経発達への影響
本症候群における神経系への影響は、患者さんの自立度や生活の質を最も大きく左右する中心的な要素です。中等度〜重度の全般性発達遅滞(GDD)と知的障害がほぼ全例に認められ、特に表出言語(しゃべる言葉)の獲得が著しく遅れる、あるいは欠如するケースが多く報告されています。
- 発達遅滞・知的障害:約95%に認められる中核症状。中等度〜重度が中心
- 言語の遅れ:表出言語の獲得が欠如、または著しく遅延
- てんかん:約50%に発症。異常な脳内ネットワーク形成が背景
- 自閉スペクトラム症(ASD):約40%。注意欠如・多動症(ADHD)の合併も多い
- セラピー負担:理学療法・作業療法・言語療法など、月平均125時間のセラピーを要する患者も
2.3 脳梁の低形成・欠損|神経画像診断の最重要所見
頭部MRIによる神経画像評価では、中枢神経系の構造的奇形が高頻度で発見されます。とくにinvdupdel(8p)患者では約80%に脳梁の低形成または完全欠損が確認されており、これが脳機能の統合不全や重度発達遅滞の主要な解剖学的要因と考えられています。
脳梁とは、右脳と左脳をつなぐ約2億本の神経線維の太い束のことです。視覚・運動・認知などの情報を左右の半球で共有するために必須の構造で、これが先天的に形成されない(または不十分な)状態を「脳梁無形成・低形成(agenesis/hypoplasia of the corpus callosum)」と呼びます。8p重複症候群、とくにinvdupdel(8p)では約80%にこの所見が認められ、重度の発達遅滞や認知障害の構造的基盤となります。
神経学的所見としては乳児期に顕著な体幹・四肢の筋緊張低下(フロッピーインファント)が認められ、吸啜力の弱さや口唇閉鎖の不全から哺乳不良・体重増加不良(Failure to thrive)を招きます。成長に伴って、低緊張は進行性の筋緊張亢進・痙性麻痺へと移行する傾向があり、運動機能の獲得に大きな障壁となります。
2.4 特徴的な顔貌(dysmorphic features)
特有の顔つきは、臨床医が本症候群を疑うための重要な手がかりとなります。複数の所見が組み合わさって現れる点が特徴です。
- 額:高く丸みを帯びた突出した額(prominent forehead)、アーチ状の眉
- 眼:両眼開離(hypertelorism)、深く窪んだ眼
- 鼻・口:幅広い鼻根、上向きの球状の鼻先、長い人中、厚く突き出した下唇
- 頭部:大頭症(macrocephaly)、口蓋裂・口唇裂、小顎症、大耳症
- 四肢:足底(とくに第1趾・第2趾の間)や手掌の深いシワ、手指の握り込み、第5指の内反、多指症・合趾症
2.5 先天性心疾患(CHD)|医学的に最も重篤な合併症の一つ
8p重複症候群における最も重篤な合併症の一つが先天性心疾患(CHD)で、約20〜25%の患者に認められます。観察される心疾患は多岐にわたり、房室中隔欠損症(AVSD)、心室中隔欠損症(VSD)、心房中隔欠損症(ASD)、肺動脈弁狭窄症、ファロー四徴症などが報告されています。
これらの心疾患の発症には、8p23.1領域のコア重複区間に位置するGATA4とSOX7という二つの転写因子の遺伝子量増加が深く関与しています。詳しいメカニズムは「3. 原因と発症メカニズム」で解説します。
2.6 筋骨格系・消化器系・その他の全身合併症
筋骨格系では患者の約60%に脊柱側弯症・後弯症の進行が認められます。重複領域が大きい症例ほど側弯症が重篤化しやすく、座位保持の困難さに加えて、胸郭変形を通じて心肺機能に二次的な悪影響を及ぼすリスクがあります。
消化器系では、生命に直結するわけではないものの、慢性かつ重度の便秘や反復する嘔吐が患者さんの生活の質を大きく損なう要因として広く認識されています。自律神経機能不全や消化管平滑筋の緊張低下が複合的に作用している結果と考えられます。このほか、眼科的異常、感音性難聴、水腎症や腎臓の未発達などの泌尿器系異常、副腎皮質機能低下症など、8p重複症候群が全人的なシステム障害であることを示す合併症が散見されます。
3. 原因と発症メカニズム|なぜ8p重複が起こるのか
8p重複症候群の症状は、重複範囲に含まれる複数の遺伝子のコピー数増加(遺伝子量効果)によって生じます。とくに8p23.1領域は、人ゲノムの中でも構造的再編成を起こしやすい「ゲノムの不安定領域(ホットスポット)」として知られており、その背景には嗅覚受容体(OR)遺伝子クラスターの存在があります。
3.1 非対立遺伝子間相同組換え(NAHR)と嗅覚受容体遺伝子クラスター
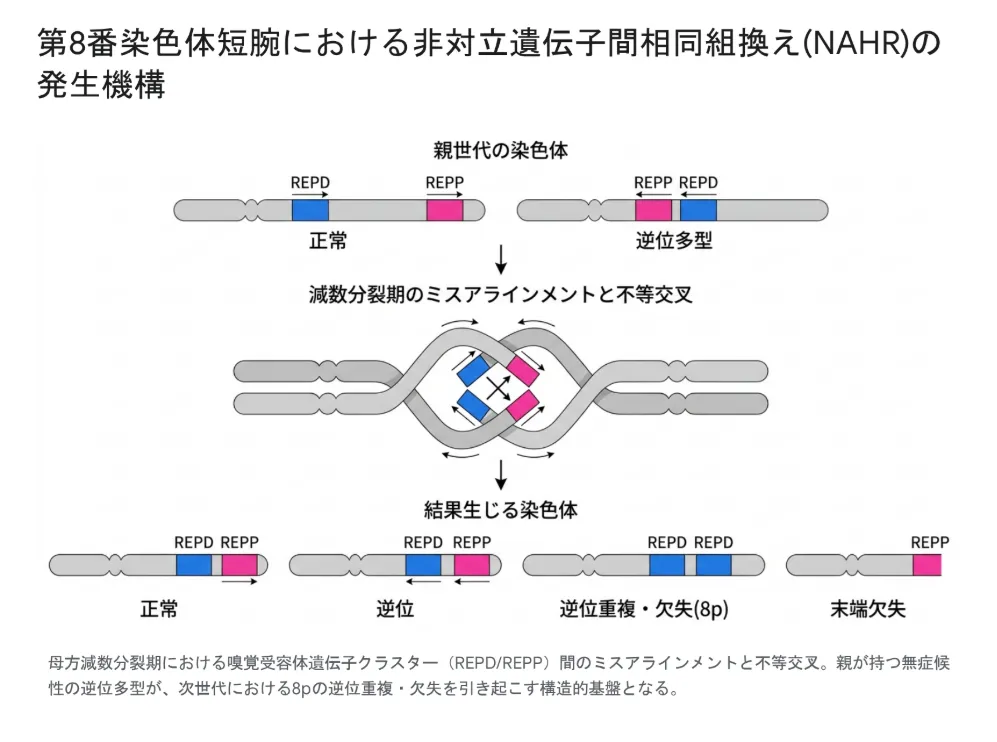
8p23.1領域の両端には、REPD(遠位)とREPP(近位)と呼ばれる巨大な反復配列(低コピー反復配列:LCR)が存在し、いずれも嗅覚受容体遺伝子クラスターから構成されています。この2つの反復配列が母方の減数分裂期に誤って対合(ミスアラインメント)し、不等交叉を起こすことで、約5Mbの領域に欠失・重複が生じます。これが「非対立遺伝子間相同組換え(NAHR)」と呼ばれる主要な発症メカニズムです。
通常、減数分裂では相同染色体(父由来と母由来の同じ番号の染色体)どうしが正しく対合し、染色体の同じ位置で組換えが起こります。しかし、染色体上にDNA配列の似た「反復配列」が複数存在すると、誤った場所どうしが対合してしまい、その結果として遺伝物質の欠失や重複が生じます。これがNAHR(Non-Allelic Homologous Recombination)です。8p23.1領域の嗅覚受容体遺伝子クラスター(REPD/REPP)は、この典型的な「足場」として機能しています。
特筆すべき疫学的背景として、ヨーロッパ系人口の約26%は、この8p23.1領域(REPDとREPPの間)に「パラセントリック逆位多型」と呼ばれるゲノム多型を有していることが知られています。この逆位自体は遺伝子量の増減を伴わないため完全に無症候性ですが、減数分裂時に相同染色体間の正常な対合を物理的に妨げ、次世代におけるNAHRの発生リスクを高める背景因子となります。つまり、両親が健康であっても、このゲノム多型を保有しているために、お子さんで重篤な8p再編成が生じうるのです。
3.2 GATA4とSOX7|先天性心疾患の鍵となる転写因子
8p23.1領域のコア重複区間(約3.68Mb・32遺伝子を含む)には、心臓発生を制御する2つの重要な転写因子GATA4とSOX7が存在します。これらの遺伝子量の変化が、先天性心疾患の発症に深く関与しています。
GATA4のハプロ不全(コピー数が1つに減ること)が心房中隔欠損や心室中隔欠損と強く結びつくことは確立されていますが、GATA4が「重複」して過剰発現した場合に単独で心疾患を引き起こすかについては長年論争がありました。1645人の発達障害児を対象とした網羅的解析で、GATA4を含む8p23.1の単独微小重複を持つ症例の中に心疾患を全く発症していないケースが見つかった一方で、GATA4の重複に加えて別の遺伝学的異常を併せ持つ患者では複雑な心疾患が観察されたのです。これは、GATA4の過剰発現が単体では決定的ではなく、他の遺伝的・環境的要因(セカンドヒット)が加わって初めて臨床的に明白な心疾患に至る「ツーヒットモデル」を強く支持する所見でした。
近年の研究では、GATA4とSOX7が「オリゴジェニックな相互作用」を形成していることが明らかになりました。マウス胚性幹細胞(ESC)を用いた心筋形成モデルでは、GATA4がSOX7の発現を直接かつ迅速に誘導し、産生されたSOX7タンパク質がGATA4やNKX2.5といった他の主要な心臓転写因子と協調して、心血管形態形成に不可欠な下流遺伝子BMP2の転写を強力に活性化することが示されています。
したがって、8p重複によってGATA4とSOX7の双方が過剰発現すると、この相互作用ネットワークが異常に駆動され、細胞内シグナルバランスが破綻し、結果として不完全な浸透率を伴う心室中隔欠損や大動脈弁異形成などの心構造異常が引き起こされると考えられています。
3.3 構造異常の多様性とブレイクポイントホットスポット
8p領域の再編成はNAHR以外にも複数のメカニズムによって起こります。invdupdel(8p)の構造において、欠失領域と重複領域の間に単一コピー(正常)の領域が存在する事実は、8pのブレイクポイント(切断点)が対称的ではないことを示しています。最近のマイクロアレイ解析により、8p内には少なくとも4つの主要な切断ホットスポット(約10.45Mb、24.32〜24.82Mb、32.19〜32.77Mb、38.94〜39.72Mb)が存在し、これらが複雑な再編成の足場となっていることが確認されています。
3.4 遺伝形式と再発リスク
・新生突然変異(de novo):両親に異常がなく、お子さんで新たに突然変異として欠失・重複が発生したケース。8p重複症候群の大半はこれに該当します。
・無症候性キャリア:ヨーロッパ系の約26%が保有する「パラセントリック逆位多型」のように、本人には症状を起こさないが、次世代で再編成のリスクを高めるゲノム多型を持つ状態。8p23.1の再編成では、このキャリアからの発生が知られています。
8p重複症候群の大半は親から遺伝したものではなく、初期胚発生における新生突然変異として発生します。したがって次のお子さんへの再発リスクは原則として低いとされています。ただし、無症候性の逆位多型を親が保有しているケースでは、次世代での再編成リスクが理論的に上昇するため、お子さんで欠失・重複が見つかった場合は両親への染色体検査を検討することが、その後の家族計画上重要なステップとなります。
4. 8p重複症候群の診断方法と鑑別診断
確定診断には染色体マイクロアレイ検査(CMA)が不可欠です。従来のGバンド染色体検査(核型分析)では微小な重複や複雑な逆位重複欠失を見逃すことが多いため、CMAを用いることが現在の診断のゴールドスタンダードとなっています。
4.1 出生後の確定診断|CMAがゴールドスタンダード
お子さんがすでに生まれており、原因不明の発達遅滞・知的障害・てんかん・特徴的な顔貌・心疾患などで医療機関を受診した場合、臨床評価で本症候群を疑い、血液検体を用いた染色体マイクロアレイ検査を行います。確定診断された場合、両親の血液で同じ重複・逆位多型の有無を確認し、頭部MRI、心エコー、腎エコー、脳波、眼科・耳鼻科診察などで合併症の精査を進めます。
CMA(chromosomal microarray analysis)は、従来のGバンド法では検出できない数kb〜数Mb単位の微小な欠失や重複(コピー数変異:CNV)を網羅的に検出する検査です。aCGHやSNPアレイなど複数の方式があり、8p重複症候群のように欠失・重複・正常コピー領域が複雑に組み合わさるケースでも、各領域のブレイクポイントを正確にマッピングできるという決定的な利点を持ちます。日本では原因不明の発達遅滞・知的障害・多発奇形に対する保険適用検査として実施されています。
4.2 検査方法ごとの比較
| 検査方法 | 特徴 | 8p重複の検出 |
|---|---|---|
| 染色体マイクロアレイ(CMA) | 確定診断のゴールドスタンダード。ブレイクポイントを正確に同定 | ◎ 確実に検出 |
| Gバンド法(核型分析) | 解像度は約5〜10Mb | ✕ 微小重複・複雑な再編成は見逃しやすい |
| FISH法 | 8p23.1領域専用プローブで迅速確認 | △ 専用プローブで可能 |
| 全エクソームシーケンス(WES) | 遺伝子の塩基配列を網羅的に解析 | △ 解析設定によりCNVも検出可能 |
4.3 鑑別診断|似た症状を示す疾患
8p重複症候群は症状が多彩なため、初期評価では他の遺伝性症候群と紛らわしいことがあります。とくに以下の疾患群との鑑別が重要です。
- 22q11.2欠失症候群(ディジョージ症候群):胎児期に円錐動脈幹奇形(ファロー四徴症など)が見つかった場合、最初に疑われがちな代表的微小欠失症候群。8p23.1領域の異常と臨床的に重なるためCMAでの鑑別が必須。
- 8p23.1欠失症候群:同じ8p23.1領域の「欠失」による疾患。GATA4のハプロ不全により先天性心疾患が高頻度で出現するが、表現型は重複型と異なる。
- その他の微小欠失・重複症候群:1p36、4p16(ウォルフ・ヒルシュホーン)、5p15(猫鳴き)、15q11.2-q13(プラダー・ウィリ/アンジェルマン)など発達遅滞を伴う症候群とCMAで鑑別。
- 純粋8p23.1重複症候群とinvdupdel(8p):同じ8p重複でも、純粋な微小重複型は脳梁欠損などの重篤な構造異常がinvdupdel(8p)より少ない傾向があり、CMAでブレイクポイントを正確に同定することが予後評価に直結。
お子さんの発達や検査結果が気になっていませんか?
原因不明の発達遅滞・特徴的な顔貌・心疾患には染色体マイクロアレイ検査が有効です。
臨床遺伝専門医にご相談ください。
※オンライン診療も対応可能です
5. 治療と長期管理|Santucci2024ガイドラインに基づく包括的アプローチ
8p重複症候群には根本的な治療法はまだ存在しません。治療は症状に応じた対症療法・外科的修復・早期療育・継続的支援が中心となります。2024年にコロラド小児病院のSantucciらが『Clinical Genetics』誌に発表した包括的レポートにより、24名の8p専門コホートに基づく画期的な臨床管理・サーベイランスガイドラインが初めて提唱されました。
5.1 「オーケストラ指揮者」モデル|多学的アプローチの実践
8p重複症候群の管理には、単一の診療科では対応できません。遺伝子専門医、小児神経科医、発達小児科医、小児循環器科医、理学療法士、作業療法士、言語聴覚士、神経心理学者、ソーシャルワーカーなど多様な専門家による緊密な連携が不可欠です。Project 8p財団は、この医療連携を「オーケストラ指揮者モデル」と呼んでいます。複雑な染色体再編成の専門知識を持つコア臨床医(遺伝専門医や小児科医)が指揮者として機能し、地域に散在する各専門医のケアを調和させることで、治療方針の衝突を防ぎます。
5.2 ライフステージ別の管理
| ライフステージ | 主な対応 |
|---|---|
| 新生児期(0〜28日) | 心エコーによる先天性心疾患の評価、筋緊張低下による哺乳不良への対応、必要に応じ経管栄養 |
| 乳児期・幼児期(〜5歳) | 早期介入プログラム(PT・OT・ST)の導入、口蓋裂・心疾患の手術、てんかんの早期管理、聴覚・視覚評価 |
| 学童期(6〜12歳) | 個別化教育計画(IEP)に準じた特別支援教育、脊柱側弯症の進行評価と装具・手術、てんかん継続管理 |
| 思春期・成人期 | 移行期医療、生活自立支援、就労支援、消化器症状(便秘・嘔吐)への継続管理、家族介護負担の軽減 |
5.3 てんかん管理と心血管系の評価
本症候群では患者の約半数がてんかんを発症するため、小児神経科医による定期的な脳波(EEG)モニタリングが必須となります。発作の種類に応じた抗てんかん薬の調整が行われ、難治性の場合は治療計画の頻繁な見直しが求められます。心血管系については、診断時に直ちに小児循環器科医による心エコー検査を行い、VSDやAVSDなどの構造的異常の有無を確認します。異常が見られる場合は、重症度に応じて定期的なフォローアップから早期の外科的修復まで包括的な管理計画が策定されます。
5.4 消化器・栄養・睡眠の最適化
TREND Communityが主導した患者データ解析では、慢性かつ重度の便秘と反復する嘔吐が、従来の医学文献の記載以上に多くの患者を悩ませていることが明らかになっています。胃食道逆流症(GERD)に対しては体位変換や経管栄養などの保存的療法から開始し、必要に応じて胃瘻造設や噴門形成術などの外科的処置が検討されます。睡眠障害に対する管理計画の策定も、家族全体の負担軽減のために重要視されています。
5.5 二重遺伝学的診断への警戒
Santucciらのコホートにおいて、患者の13%(24名中3名)が8pの異常に加えてゲノム上の別領域にも病原性変異を持つ「二重遺伝学的診断」を受けていた事実が報告されています。例えば、ある患者は8p23.3-23.1の末端欠失に加えて4p16.3領域に4Mbの重複を有していました。8pの一般的な表現型とは異なる非典型的な症状(極端な成長障害や特異な代謝異常など)が現れた場合は、8p異常のみに固執せず、ゲノム全体の追加評価を考慮することが推奨されています。
5.6 未来の治療戦略|Project 8p研究ロードマップ
現代の医学では8p異常を根本的に治療する手段は存在しませんが、革新的な研究が前例のないスピードで進行中です。Project 8p Foundationは、iPSC(人工多能性幹細胞)と大脳オルガノイドによる疾患モデリング、ゲノム編集技術を応用した「染色体工学」によるダイソミー(二倍体)回復戦略、Insight Portal・CNV Portalによるオープンサイエンス、TeleEchoモデルによる遠隔医療教育など、多角的なアプローチを並行して推進しています。数年後には基礎研究の成果が臨床試験へと移行し、患者さんに直接的な利益をもたらすことが期待されています。
6. 遺伝カウンセリングと再発リスク
8p重複症候群は表現型の幅が広く、予後予測が容易ではありません。遺伝カウンセリングを通じて、ご家族が病気を正確に理解し、納得のいく決断ができるよう中立的な情報提供を行うことが、医師の重要な役割です。
6.1 カウンセリングで伝えるべきポイント
- 重複範囲と症状の関係:含まれる遺伝子(GATA4・SOX7など)によって症状の重症度が変わる
- サブタイプによる予後の違い:invdupdel(8p)・純粋微小重複・モザイク型・環状染色体で大きく異なる
- 表現型の多様性:軽症から重症まで幅広いスペクトラム。同じ重複でも経過は個人ごとに異なる
- 両親の検査:新生突然変異か逆位多型キャリアからの発生かを判定し、次世代のリスク評価につなげる
- 支援体制:多職種チーム、早期療育、社会福祉制度、Project 8p Foundationなど国際的な家族会の紹介
6.2 再発リスクの考え方
| 状況 | 次子への再発リスク |
|---|---|
| 両親とも異常なし(新生突然変異) | 原則として低い(1%未満)※生殖細胞モザイクの可能性は残る |
| 親が8p23.1パラセントリック逆位多型キャリア | 理論的に上昇(NAHR発生リスクが高まる) |
| 親が均衡型染色体転座保有 | 転座の種類によりリスクが異なる(個別評価が必要) |
7. 出生前診断とミネルバクリニックのサポート体制
8p重複症候群は、NIPTのうち全染色体スクリーニング型のプラン(インペリアルプラン)でリスクを評価でき、羊水検査・絨毛検査でCMAを行うことで確定診断が可能です。ただし、出生前に見つけることが常にご家族の利益になるとは限らないため、検査前後の遺伝カウンセリングが不可欠です。
7.1 超音波検査で疑われる胎児所見
8p重複・逆位重複欠失症候群を決定づける単一の特異的マーカーは超音波検査には存在しませんが、形態形成の異常を反映する複数のソフトマーカーが観察されることがあります。代表的な所見としては、妊娠初期の後頸部浮腫(NT)肥厚、妊娠中期の側脳室拡大、頭蓋顔面の異形症、心臓・腎臓の構造異常などが挙げられます。実際に、心構造異常(VSD・大動脈弓狭窄など)を契機に22q11.2欠失症候群が疑われたものの、その後の羊水検査で8p23.1の重複と8p23.2の異常な増幅が同定された症例も報告されており、超音波所見が必ずしも一般的な染色体異常に起因するわけではないことを臨床医に示しています。
7.2 出生前検査の種類と検出能力
| 検査 | 位置づけ | 8p重複への対応 |
|---|---|---|
| NIPT(一般的なターゲット型) | スクリーニング検査 | 対応していないことが多い(特定の微小欠失のみを対象とするプランでは対象外) |
| NIPT(全染色体スクリーニング型) | スクリーニング検査 | ○ スクリーニング可能(5Mb以上を対象とするWGS型では8p23.1領域もカバー) |
| 絨毛検査+CMA | 確定診断 | ◎ 妊娠初期に確定診断可能 |
| 羊水検査+CMA | 確定診断 | ◎ 微小重複・逆位重複欠失も確定診断 |
7.3 ミネルバクリニックでのNIPTプラン
ミネルバクリニックでは、ご家族のニーズに応じて複数のNIPTプランをご用意しています。ダイヤモンドプランはターゲット法による高精度検査で、特定12箇所の微小欠失(1p36、4p16、5p15、9p、22q11.2など)を高い陽性的中率で検出しますが、8p重複はこの12箇所には含まれません。一方インペリアルプランはWGS法とターゲット法のハイブリッドで、5Mb以上の全染色体微小欠失・重複を広範囲にスクリーニングするため、8p23.1重複症候群・8p重複もカバーされます。スクリーニング検査のため、陽性時は羊水検査・絨毛検査による確定診断が必要です。
7.4 出生前診断で見つかった場合の対応
出生前に8p重複が見つかった場合、本症候群は表現型の幅が非常に広いため、胎児期の超音波所見だけで将来の予後を正確に予測することは難しい場合があります。とくに「意義不明なバリアント(VUS)」として報告されるケースでは、両親は重篤な超音波所見と不確実な遺伝学的予後の間で、極めて困難な意思決定を迫られます。遺伝カウンセリングで重複範囲・関与する遺伝子・表現型の幅・予後の不確実性を中立的に説明し、両親の検査で新生突然変異か逆位多型キャリアからの発生かを判定し、詳細超音波で心奇形・脳梁の構造・四肢異常などを精査します。心疾患が疑われる場合はNICUを備えた高次医療機関での出産を検討し、ご家族の不安や葛藤に寄り添い、決断を急がせない時間と環境を確保することが何より大切です。
⚖️ 倫理的なスタンス|検査は「常に利益」ではない
8p重複症候群のように不完全浸透や表現型の幅が大きい疾患では、出生前に見つけたことが必ずしもご家族の利益になるとは限りません。「特定の検査を勧める」「安心を保証する」「不安をあおる」ような表現は適切ではないと私たちは考えています。検査を受けるかどうか、結果をどう受け止めるかは、十分な情報を得たうえで、ご家族自身が決めるべき事柄です。
7.5 ミネルバクリニックのサポート体制
ミネルバクリニックでは、臨床遺伝専門医の専門性を活かした診療体制を整えています。8p重複症候群を含む染色体微小欠失・重複症候群について、出生前検査から結果説明、確定検査、その後のフォローまで一貫してサポートいたします。
- 全染色体スクリーニング対応:インペリアルプランでは5Mb以上の全染色体微小欠失・重複を広くスクリーニング、8p23.1領域・8p重複もカバー対象
- 確定検査も院内で実施:羊水検査・絨毛検査も院内で実施可能、転院の必要なし
- 臨床遺伝専門医が担当:臨床遺伝専門医が検査前後の遺伝カウンセリングを直接担当
- 互助会で費用面も安心:NIPT受検者全員に適用される互助会(8,000円)により、陽性時の羊水検査費用が全額補助
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
よくある質問(FAQ)
関連記事
参考文献
- Santucci K, Malik KE, Angione K, et al. Chromosome 8p Syndromes Clinical Presentation and Management Guidelines. Clin Genet. 2025;107(2):169-178. [PubMed]
- Okur V, Hamm L, Kavus H, et al. Clinical and Genomic Characterization of 8p Cytogenomic Disorders. Genet Med. 2021;23(12):2342-2351. [PubMed]
- Vibert R, Mignot C, Keren B, et al. Neurodevelopmental Phenotype in 36 New Patients With 8p Inverted Duplication-Deletion: Genotype-Phenotype Correlation for Anomalies of the Corpus Callosum. Clin Genet. 2022;101(3):307-316. [PubMed]
- Giglio S, Broman KW, Matsumoto N, et al. Olfactory Receptor-Gene Clusters, Genomic-Inversion Polymorphisms, and Common Chromosome Rearrangements. Am J Hum Genet. 2001;68(4):874-883. [PubMed]
- Barber JC, Hall V, Maloney VK, et al. 8p23.1 duplication syndrome: narrowing of critical interval to 1.80 Mbp. Mol Cytogenet. 2013;6(1):28. [PMC]
- Glancy M, Barnicoat A, Vijeratnam R, et al. Cardiac defects are infrequent findings in individuals with 8p23.1 genomic duplications containing GATA4. Circ Cardiovasc Genet. 2011;4(4):349-357. [PubMed]
- Orphanet – 8p inverted duplication/deletion syndrome (ORPHA:96092) [外部サイトへ]
- Orphanet – 8p23.1 duplication syndrome (ORPHA:251076) [外部サイトへ]
- Orphanet – Trisomy 8p syndrome (ORPHA:264450) [外部サイトへ]
- GARD – 8p23.1 duplication syndrome [外部サイトへ]
- GARD – Partial duplication of the short arm of chromosome 8 [外部サイトへ]
- Unique – Rare Chromosome Disorder Support Group: 8p duplications [外部サイトへ]
- Unique – 8p inverted duplication & deletion [外部サイトへ]
- Project 8p Foundation – Researchers Roadmap [外部サイトへ]
- MedlinePlus Genetics – Chromosome 8 [外部サイトへ]






