目次

📍 クイックナビゲーション
ヒトのゲノム構造において、各染色体の構造的な完全性と遺伝子の発現量(ドセージ)の厳密なコントロールは、正常な発生と生命維持に不可欠です。その中でも「11番染色体」は非常に遺伝子密度が高く、少しの異常が全身の多臓器に甚大な影響を及ぼすことがわかっています。
本記事では、11番染色体が引き起こす「トリソミー(過剰)」や「微小欠失(一部の欠け)」、「微小重複(一部の過剰)」といった多様な染色体異常について、最新の分子遺伝学の知見に基づいて、臨床遺伝専門医が網羅的かつわかりやすく解説します。
1. 11番染色体とは?ゲノム構造と重要な役割
ヒトの11番染色体は、約1億3400万塩基対(134 Mb)から構成されており、ヒトゲノム全体の約4〜4.5%を占めています。物理的な構造として、短いほうの腕である短腕(11p)は約50 Mb、長いほうの腕である長腕(11q)は約84 Mbの長さを持ちます。
この染色体には全体で約1,500個もの遺伝子がマッピングされており、遺伝子密度の高さが特徴です。これら1,500個の遺伝子のうち、少なくとも約200個の遺伝子の変異や量の異常が、先天性の病気や障害の直接的な原因になることがわかっています。
📚 医学用語解説:常染色体における「顕性」と「潜性」
11番染色体のような性染色体以外の染色体を「常染色体」と呼びます。遺伝の仕方には、片方の遺伝子に変異があるだけで発症する常染色体優性(顕性:けんせい)遺伝と、両方の遺伝子に変異が揃わないと発症しない常染色体劣性(潜性:せんせい)遺伝があります。※現在、医学会では優劣の誤解を避けるため「顕性・潜性」という言葉への移行が進んでいます。
以下の図は、11番染色体の構造と、微小欠失や微小重複によって引き起こされる主要な疾患が、染色体上のどの領域(番地)に対応しているかを示したマップです。
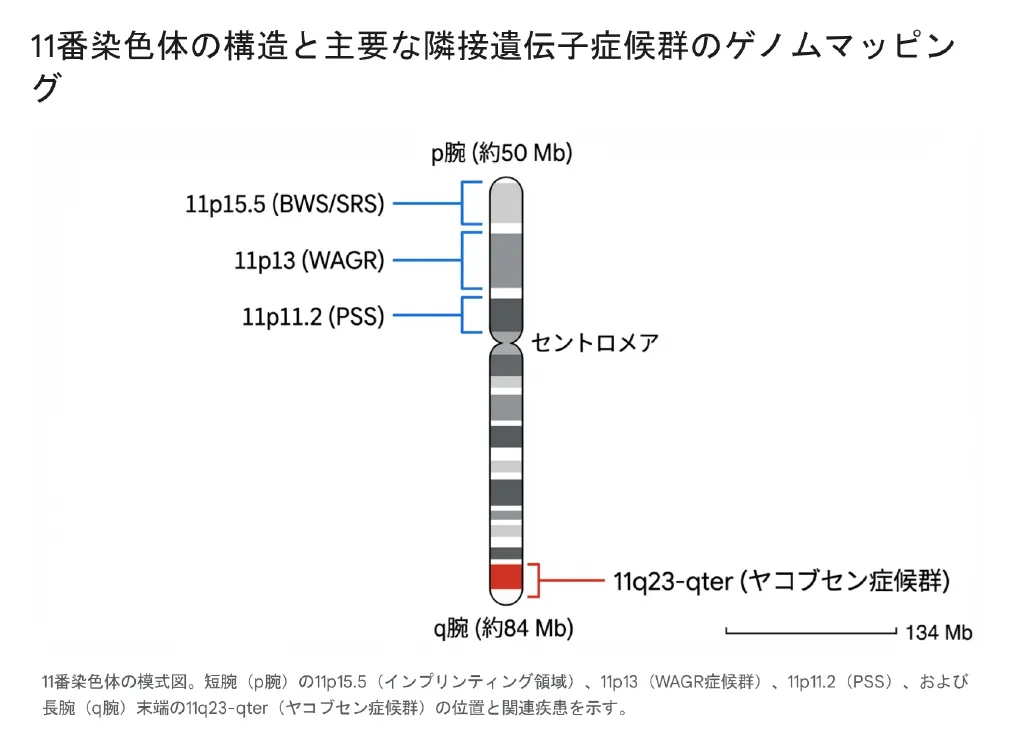
このように、11番染色体は少しでも構造が変化する(重複する、あるいは欠失する)と、その位置に応じて全く異なる複雑な症候群を引き起こします。これまでに世界で1,400人以上の患者が何らかの構造異常を有していると報告されています。
2. 11番染色体の数的異常:完全トリソミーとモザイクトリソミー
完全な「トリソミー11」はなぜ産まれてこないのか
通常、染色体は父親から1本、母親から1本の合計2本(ペア)で構成されますが、これが3本になってしまう状態をトリソミーと呼びます。ダウン症(21番染色体トリソミー)などが有名ですが、11番染色体においては、体のすべての細胞で染色体が3本存在する「完全な11番染色体トリソミー」の赤ちゃんが無事に産まれたという報告は、医学文献上存在しません。
理由は、11番染色体に存在する多数の発生制御遺伝子や細胞周期関連遺伝子の量が正常の1.5倍に増えてしまうことで、受精卵から細胞分裂していく初期のプロセスが致命的に破綻してしまうからです。その結果、極めて早い妊娠初期の段階で自然流産に至ります。
一部の細胞だけが異常な「モザイクトリソミー」の不思議な転帰
一方で、正常な細胞(2本)とトリソミー細胞(3本)が体内に混ざって存在する「モザイクトリソミー11」は、出生前検査(NIPTや羊水検査)で発見され、無事に生まれてくるケースがあります。
🧬 医学用語解説:トリソミーレスキューと胎盤限局性モザイク(CPM)
受精卵が3本の染色体を持ってしまった場合、細胞が生き残るために自己修復を試み、分裂の途中で余分な1本を捨てる現象をトリソミーレスキューと呼びます。
この修復の結果、赤ちゃんの体を作る細胞はすべて正常(2本)に戻り、胎盤を作る細胞にだけトリソミー(3本)の細胞が残ることがあります。これを胎盤限局性モザイク(CPM)と呼び、出生前診断で異常が出ても、赤ちゃん自身は全く健康に生まれてくることが多い最大の理由です。
実際、出生前診断でモザイクトリソミー11が確認された赤ちゃんの「ほぼすべて」が、出生後に身体的・精神運動発達において完全に正常な成長を遂げています。不思議なことに、大規模な研究では、母親の年齢が高い(高齢妊娠)ほど、このモザイクにおいて正常な転帰(健康な出産)をたどる割合が高いことも判明しています。
見逃してはいけない「片親ダイソミー(UPD)」の除外
しかし、モザイクトリソミー11が発見された場合、臨床プロトコルとして絶対に確認しなければならない落とし穴があります。それが「片親ダイソミー(UPD)」です。
⚠️ 医学用語解説:片親ダイソミー(UPD)とは
トリソミーレスキューで余分な1本を捨てて2本に戻る際、運悪く「父親から来た1本」と「母親から来た1本」ではなく、「父親から来た2本」あるいは「母親から来た2本」だけが残ってしまう確率が理論上約33%あります。染色体の数は2本で正常に見えますが、遺伝子が「片親由来」に偏るため、重大な発育障害を引き起こします。
11番染色体においてこのUPDが発生すると、後述するインプリンティング異常により、巨体を伴うベックウィズ・ヴィーデマン症候群や、重度の発育不全を伴うシルバー・ラッセル症候群を発症します。そのため、マイクロアレイ検査などでUPDを確実に除外することが不可欠です。
3. 11番染色体短腕(11p)の部分トリソミーと相反する成長異常
染色体の一部が過剰になる「部分トリソミー」の中でも、11番染色体の短腕の先端にある「11p15.5領域」の異常は、分子遺伝学的に極めて特異な現象を引き起こします。
💡 医学用語解説:ゲノムインプリンティング(刷り込み)
人間は父親と母親から同じ遺伝子を1つずつ受け継ぎますが、特定の遺伝子においては「父親由来のものしか働かない」「母親由来のものしか働かない」というように、エピジェネティックなスイッチ(DNAのメチル化など)によって発現が制御されています。これをゲノムインプリンティングと呼びます。11p15.5領域は、このインプリンティングを受ける成長制御遺伝子の巨大な密集地帯です。
11p15.5領域には、強力な成長促進因子(IGF2:父親由来のみ働く)と、成長抑制因子(CDKN1C・H19:母親由来のみ働く)が存在します。
この領域が重複して「遺伝子が過剰」になった場合、その重複部分が「父親から来たか、母親から来たか」によって、鏡文字のように完全に相反する症状が現れます。
父親由来の重複:ベックウィズ・ヴィーデマン症候群(BWS)
成長促進因子(IGF2)が病的に増加し、深刻な過成長症候群を引き起こします。
主な症状:出生時の著しい過体重と過身長(巨体)、大舌症、臍帯ヘルニアなどの腹壁欠損、身体の左右非対称な過成長(片側肥大)、高インスリン血症による新生児低血糖。
最大の懸念事項として、ウィルムス腫瘍(腎芽腫)や肝芽腫などの胎児性腫瘍の発生リスクが小児期を通じて著しく増大します。
関連ページ:ベックウィズ・ヴィーデマン症候群とは?
母親由来の重複:シルバー・ラッセル症候群(SRS)
成長抑制因子(CDKN1CやH19)が過剰に働き、重度の成長障害症候群を発症します。
主な症状:出生前からの重度の子宮内発育遅延(IUGR)、出生後の持続的な低身長、摂食障害。頭囲は正常に近いものの体が極端に小さいため頭が大きく見える「相対的巨頭」と、特徴的な三角形の顔つきを呈します。
関連ページ:11p15微小重複症候群
4. 11番染色体長腕(11q)の部分トリソミーとエマヌエル症候群
11番染色体の長腕側の末端に近い領域の重複は「遠位トリソミー11q」と呼ばれ、頭蓋顔面異形態や知的障害、心房中隔欠損症などの心疾患を引き起こします。中でも非常に特異で複雑なのが「エマヌエル症候群」です。
エマヌエル症候群:2つの染色体が複合する特殊な病態
エマヌエル症候群は、全ての染色体異常症候群の中で唯一、「11番染色体と22番染色体の両方の部分トリソミーが合体して発生する」という稀有なメカニズムを持ちます。
発症メカニズムは細胞遺伝学的に非常に特殊です。患者の99%以上において、両親のどちらかが11番と22番の染色体の一部が入れ替わった「均衡型転座 t(11;22)」を持っています。親自身は遺伝情報の総量に過不足がないため完全に無症状ですが、生殖細胞を作る際の特殊な染色体分配エラー(3:1分離)により、次世代の受精卵に「過剰な派生染色体22」が受け継がれます。
これにより、11qの遠位部分と22qの近位部分の遺伝子を通常の2コピーではなく「3コピー」持つことになり、重度の知的障害、顕著な小頭症、口蓋裂、生存を脅かす重症先天性心疾患や腎奇形など、多臓器にわたる深刻な症状を引き起こします。
5. 11番染色体短腕(11p)の微小欠失:WAGR・PSS
染色体の一部が「欠失する(失われる)」ことで発生するのが部分モノソミー(微小欠失症候群)です。11番染色体は遺伝子が密集しているため、少し欠けただけでも複数の重要な遺伝子が同時に失われ、深刻な隣接遺伝子欠失症候群を引き起こします。
🔍 医学用語解説:ハプロ不全
通常、遺伝子は2つ1組で働くことで必要な量のタンパク質を作り出します。片方の遺伝子が失われる(欠失する)ことで、作られるタンパク質の量が半減してしまい、正常な身体機能や臓器の形成が維持できなくなる状態をハプロ不全と呼びます。微小欠失症候群の根本的な原因です。
WAGR症候群(11p13微小欠失)
11番染色体短腕の「11p13」領域の微小欠失によって引き起こされる稀な多発先天奇形および腫瘍好発症候群です。疾患名は、主要な4つの症状の頭文字から命名されています。
- W:ウィルムス腫瘍(Wilms Tumor):患者の45〜60%という極めて高い確率で発生する小児期の悪性腎臓がん。強力な腫瘍抑制遺伝子であるWT1遺伝子の欠失が原因です。
- A:無虹彩症(Aniridia):眼球の虹彩の完全または部分的な欠損。PAX6遺伝子の欠失によるもので、重度の視力低下を伴います。
- G:泌尿生殖器異常(Genitourinary anomalies):停留精巣、尿道下裂、女性における索状生殖腺など。
- R:精神発達遅延(Retardation/Range of developmental delays):知的障害やADHD、自閉症スペクトラム障害などの神経発達障害。
さらに、近接するBDNF遺伝子まで欠失が及ぶと、制御不能な過食と重度の肥満を伴うWAGRO症候群となります。臨床管理として、腫瘍の急速な成長に備え、生後から7歳までは3ヶ月ごとの厳格な腹部超音波検査が強く推奨されています。
関連ページ:11p13(WAGR症候群)
ポトツキー・シェイファー症候群(PSS:11p11.2微小欠失)
医学文献上でも報告が非常に少ない、11p11.2領域の欠失症候群です。欠失した個々の遺伝子が、それぞれ明確で特異的な症状を独立して引き起こすのが特徴です。
- EXT2遺伝子:良性の骨腫瘍が多発する多発性外骨腫を引き起こします。
- ALX4遺伝子:頭蓋骨の骨化が遅延し、頭頂部に異常に拡大した「頭頂孔」を生涯にわたって残します。
- PHF21A遺伝子:重度の知的障害や、特異な顔貌(幅広く短い頭蓋骨、短い人中など)、乳児期早期の難治性てんかん(点頭てんかん)の主要な原因となります。
関連領域ページ:11p11.2
EXT2遺伝子等の関連検査:遺伝性多発性骨軟骨腫(遺伝性多発性外骨腫症)NGS遺伝子検査パネル|ミネルバクリニック
6. 11番染色体長腕(11q)の末端欠失:ヤコブセン症候群
11番染色体長腕の末端部(11q23.3からテロメアに至る領域)の欠失によって引き起こされる重篤な疾患が「ヤコブセン症候群」です。女性患者が男性の2倍多いという特徴があります。染色体の切断が起きやすい脆弱部位(FRA11B)が欠失の起点となることが多いです。
この症候群の予後を決定づける重篤な合併症は以下の通りです。
- 血液異常(パリ・トルーソー症候群):ほぼ100%の患者が合併する出血性疾患。FLI1遺伝子の欠失により血小板の造血プロセスが阻害され、生涯を通じて内出血や致死的な頭蓋内出血のリスクを抱えます。
- 先天性心疾患(CHD):患者の半数以上が発症。ETS1遺伝子のハプロ不全により、特に左心低形成症候群(HLHS)などの左心系病変が極めて高頻度で発生します。
- 頭蓋顔面異常と神経発達障害:前頭縫合の早期癒合による「三角頭蓋」、広範な消化器・泌尿器奇形、そして全例において知的障害や高い有病率の自閉症スペクトラム障害(ASD)が認められます。
ヤコブセン症候群の生存・予後は、重度の心臓外科手術に耐えられるか(出血リスク・麻酔リスクとの闘い)に大きく依存します。
関連ページ:
11q24.1(ヤコブセン症候群関連領域)
ヤコブセン症候群の責任領域とMCR解析(11q23q25)
7. 最新の遺伝子検査技術と、未来の精密医療(Precision Medicine)
11番染色体の構造異常が引き起こす表現型の複雑さを考えると、迅速かつ超高精度な遺伝子診断が不可欠です。
かつての顕微鏡での核型分析(Gバンド法)では、WAGR症候群などの数Mb以下の微細な欠失を見つけることは困難でした。しかし現在では、ゲノム全体のコピー数変異を高解像度でスキャンする染色体マイクロアレイ解析(CMA)や、次世代シーケンシング(NGS)を応用した中深度全ゲノムシーケンス(CMA-seq)が導入され、出生前・生後における診断精度は飛躍的に向上しています。
現在、これらの11番染色体異常に対して、欠損した遺伝子自体を修復するような根治的治療は存在せず、各専門医の連携による対症療法と生涯のモニタリングに依存しています。
しかし、基礎医学の領域では画期的な進展が起きています。他の染色体異常(15番染色体のアンジェルマン症候群など)において、アデノ随伴ウイルス(AAV)ベクターを用いた遺伝子補充療法や、CRISPR-Casを利用した遺伝子編集治療の臨床試験が急速に進行しています。11番染色体の微小欠失に起因するハプロ不全に対しても、将来的にはゲノム編集療法やアンチセンスオリゴヌクレオチド(ASO)療法が適用可能になるという、ゲノム医療の大きな希望が見え始めています。
ミネルバクリニックでは、最新のNGS技術を用いた網羅的な遺伝子検査を提供しています。詳細はこちらをご覧ください。
ミネルバクリニック取り扱い遺伝子検査一覧
よくある質問(FAQ)
関連記事
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
参考文献
- Chromosome 11 Introduction: The genetic size of chromosome 11. [外部資料]
- The Genetics of Microdeletion and Microduplication Syndromes: An Update. PMC-NIH. [PMC]
- Mosaicism for Autosomal Trisomies: A Comprehensive Analysis of 1266 Published Cases Focusing on Maternal Age and Reproductive History. [MDPI]
- Beckwith-Wiedemann syndrome/spectrum. Children’s Hospital of Philadelphia. [CHOP]
- Silver-Russell Syndrome and Beckwith-Wiedemann Syndrome: Opposite Phenotypes with Heterogeneous Molecular Etiology. [PMC]
- Emanuel Syndrome – GeneReviews® – NCBI Bookshelf. [NCBI]
- WAGR Syndrome Spectrum Disorder – GeneReviews®. [NCBI]
- Potocki-Shaffer syndrome – Genetics – MedlinePlus. [MedlinePlus]
- Jacobsen Syndrome (11q Terminal Deletion Syndrome) – StatPearls – NCBI. [NCBI]
- CRISPR Clinical Trials: A 2025 Update – Innovative Genomics Institute (IGI). [IGI]






