目次

📍 クイックナビゲーション
第16番染色体は、細胞遺伝学および周産期医療において極めて特異かつ重要な位置を占める染色体です。全体のおよそ90メガベース(Mb)のDNAペアから構成され、比較的小型でありながら極めて高い遺伝子密度を有しています。本記事では、16番染色体に生じる多様なゲノム異常について、臨床遺伝専門医の視点から最新データを交えて徹底解説します。
Q. 16番染色体異常にはどのようなものがありますか?
A. 全ての細胞が異常となる完全トリソミーから、微細な欠失まで幅広いスペクトラムが存在します。
完全トリソミーは胎児期に致命的となりますが、正常細胞が混ざる「モザイク型」や、染色体の一部だけが増減する「微小欠失・重複」の場合は出生可能であり、それぞれ全く異なる症状や予後を辿ります。
- ➤完全トリソミーと流産 → 全自然流産の約6〜7.5%を占める最も頻度の高い異常です。
- ➤モザイクトリソミーのパラドックス → 周産期リスクは高いものの、長期的な生活の質(QOL)は予想以上に良好です。
- ➤短腕(16p)の微小欠失・重複 → 自閉症や肥満と関連する16p11.2異常などについて。
- ➤長腕(16q)の異常 → 非常に深刻な表現型を示す部分トリソミーやACD/MPVについて。
1. 16番染色体異常の全体像と完全トリソミー
ヒトの初期発生において、16番染色体は生命の維持に直結する重要な役割を担っています。そのため、染色体全体が3本になってしまう「完全なトリソミー(Full Trisomy 16)」は、生物学的に生存が不可能(Incompatible with life)な病態とされています。
💡 重要な事実:流産の最も一般的な原因
16番染色体の完全なトリソミーは、受胎時に生じる常染色体異常の中で最も頻度が高く、全妊娠の約1〜1.5%を占めると推定されています。また、全ての自然流産の約6〜7.5%の要因となっており、大部分が最終月経後8週から15週の間に流産に至ります。
この顕著な発生致死性は、16番染色体が極めて高い遺伝子密度を有していることに起因します。これらの遺伝子が過剰に発現すること(ドーズ効果)で、致死的な細胞増殖異常が起こり、口蓋裂、重度の脳奇形(小頭症など)、重篤な先天性心疾患(心室中隔欠損)、横隔膜ヘルニアといった決定的な組織形成の破綻を引き起こすためです。
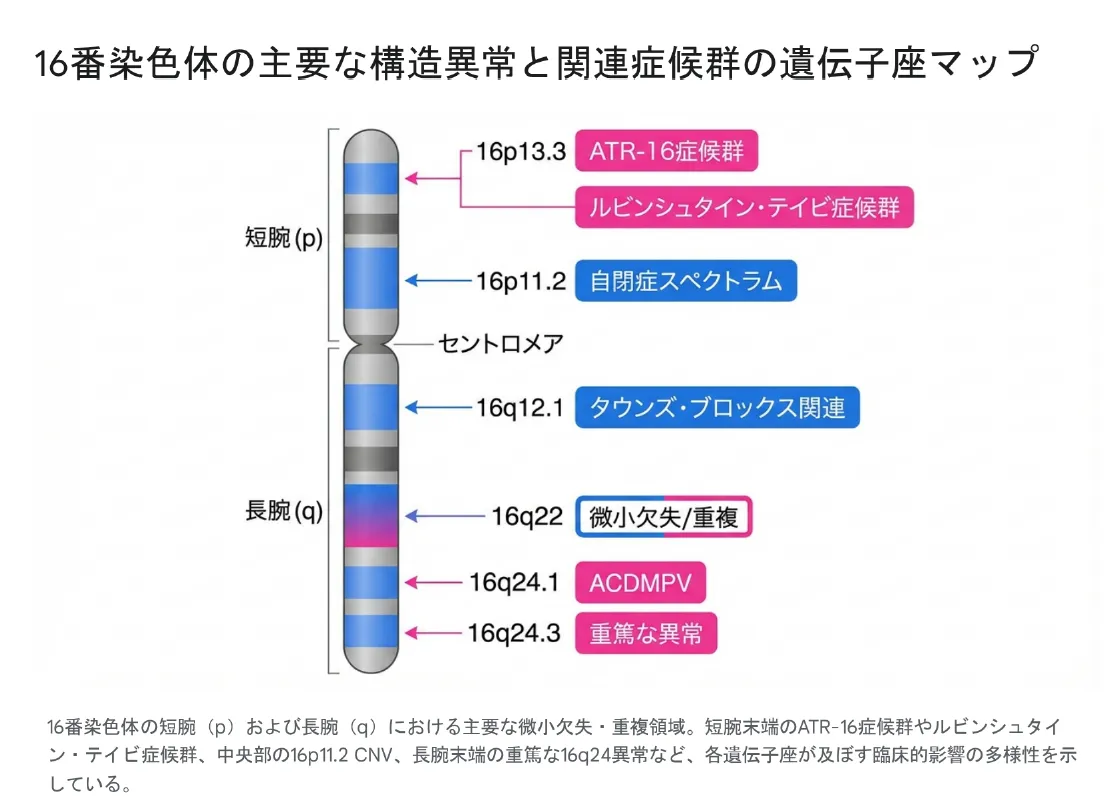
▲ 16番染色体の主要な構造異常と関連症候群の遺伝子座マップ。短腕(p)から長腕(q)にかけて多彩な疾患が紐づいている。
2. モザイクトリソミー16(MT16)と限局性胎盤モザイク(CPM)
完全トリソミーが絶対的に致死的である一方で、正常な核型の細胞と、トリソミーの細胞が同一の個体内に混在する「モザイクトリソミー16(Mosaic Trisomy 16: MT16)」の場合は、妊娠を維持して出生に至り、成人期まで生存する可能性があります。
ここでNIPT(非侵襲的出生前検査)を受ける妊婦さんに非常に重要となるのが、「限局性胎盤モザイク(Confined Placental Mosaicism: CPM)」という概念です。
🔍 専門用語解説:限局性胎盤モザイク(CPM)とは?
トリソミーなどの異常な細胞が「胎盤(絨毛)組織」にのみ存在し、胎児本体の細胞は正常である状態を指します。NIPTは主に胎盤由来のDNAを解析するため、NIPTで「16番トリソミー陽性」となっても、その後の羊水検査で「胎児は正常」と判明するケースの多くがこのCPMに該当します。
しかし、胎児が正常細胞であっても、胎盤にトリソミー細胞が混在していると胎盤機能不全に直結します。母体と胎児の栄養供給が阻害されるため、子宮内胎児発育遅延(IUGR)や妊娠高血圧腎症、自然早産のリスクが劇的に上昇します。
モザイク型の長期的な「生活の質(QOL)」のパラドックス
産科医からNIPT陽性やIUGRを指摘されたご家族にとって、最も気がかりなのは「生まれてきた後、どのような生活になるのか」という点でしょう。
最新の国際レジストリを通じた44家族の調査では、極めて高い周産期リスクと、予想をはるかに上回る良好な長期的予後のコントラストが明らかになっています。
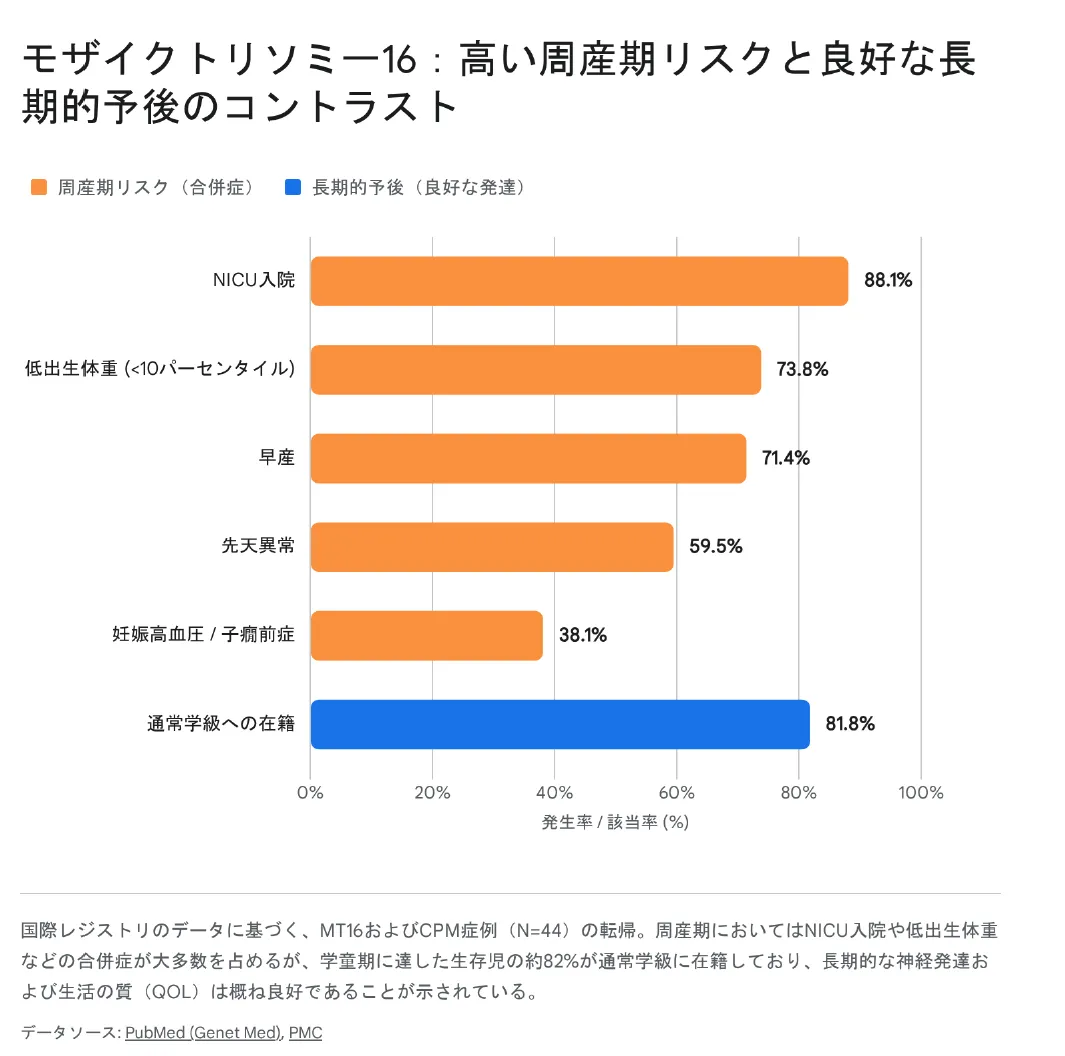
▲ グラフ:NICU入院率や低出生体重率が高い一方で、学童期に通常学級へ在籍する割合は約82%に達する。
調査対象の生存児のうち、NICU(新生児集中治療室)への入院は88.1%、早産は71.4%と極めて過酷なスタートを切ります。しかし、この時期を乗り越えて学童期に達した子どもの81.8%が、特別支援教育を必要とせず通常の学級(メインストリーム)に在籍しています。
また、生活の質(QOL)を評価する標準的指標においても、身体的・心理社会的に非常に高い水準を示しており、認知・知的障害の不可逆的な発生率は決して高くないことが裏付けられています。
3. 16番染色体短腕(16p)の微小重複(部分トリソミー)
近年、マイクロアレイ染色体検査(CMA)の普及により、従来のGバンド分染法(顕微鏡での観察)では見逃されていた極めて微小な染色体の増減(コピー数バリアント:CNV)が次々と明らかになっています。
16番染色体の短腕(16p)には、DNA配列が反復する領域が多く、細胞分裂時のエラーにより微小な重複や欠失が頻発しやすい構造的特徴があります。
16p11.2重複症候群:不完全浸透という複雑な性質
約593キロベース(kb)の領域が過剰になる16p11.2重複症候群は、自閉症スペクトラム障害(ASD)、言語発達遅滞、注意欠陥・多動性障害(ADHD)、そして思春期以降における統合失調症などの精神疾患の発症リスクを顕著に増加させます。
🧬 専門用語解説:不完全浸透(Incomplete penetrance)
この重複の最大の特徴は「持っているからといって必ず発症するわけではない」という点です。同一の重複を持つ家系内であっても、重度の自閉症を呈する子供から、全く無症状で通常の社会生活を送っている親まで、症状の現れ方が大きく異なります。
その他、16p11.2-p12.2重複症候群などの近接領域の異常も存在します。
16p13.11微小重複症候群:過度な不安は禁物
16p13.11反復性微細重複症候群もまた、行動異常や自閉症スペクトラムが報告されているものの、疾患浸透率は約7%〜10.6%と極めて低く、無症状の親から遺伝するケースが頻繁に見られます。
出生前診断でこの重複が偶然発見された場合、超音波検査で重大な心疾患や脳奇形がなければ、正常な発育を遂げるケースも非常に多いため、これを「絶対的な疾患原因」と思い込まない配慮が必要です。
🔍 関連記事:16番染色体短腕の微小重複・欠失に関する詳細はこちら
4. 16番染色体短腕(16p)の微小欠失(部分モノソミー)
ゲノムの異常において、一般的な原則として「欠失(情報の喪失)」は「重複(情報の過剰)」よりも重篤な表現型をもたらす傾向があります。この現象は「ハプロ不全(Haploinsufficiency:片方の遺伝子が失われることで十分なタンパク質が作れず病気になること)」と呼ばれます。
16p11.2微小欠失症候群:神経発達と代謝の交差点
16p11.2-p12.2欠失症候群(主に16p11.2欠失)は、自閉症全体の原因の約0.5%を占めるとされる強力な神経発達障害の要因です。
この欠失の臨床所見は非常に特徴的で、重複例とは明確なコントラスト(逆の症状)をなします。
- ➤肥満と大頭症:重複例が「低体重・小頭症」になるのに対し、欠失例は幼少期から極めて高確率(約75%)で著しい肥満と大頭症を発症します。
- ➤言語と運動の重篤な障害:約80〜90%で言語障害が見られ、小児期発症の音声運動障害が高頻度で発生します。
- ➤成人期の管理:過食や代謝異常による2型糖尿病などの二次的合併症の管理が、長期的QOLの鍵となります。
その他の重要な16p欠失症候群
■ ルビンシュタイン・テイビ症候群(RSTS)と16p13.3微細欠失症候群
CREBBP遺伝子の変異、または広範な欠失により引き起こされる常染色体顕性(優性)遺伝疾患です。重度の精神運動発達遅延、特徴的な顔貌、幅の広い特異な親指が特徴です。広範な欠失を伴う「重症型」では難治性てんかんや免疫不全を伴い生命予後が悪化します。
■ ATR-16症候群とは?
16p13.3のさらに末端(テロメア側)の欠失により、アルファグロビン遺伝子群が失われることで発症します。血液学的にアルファ・サラセミア(貧血)を呈するほか、中等度の知的障害や内反足などを合併します。
その他、16p12.1微細欠失症候群や16p12.2微細欠失症候群、16p13.11微小欠失症候群など、欠失するバンドの位置によって多彩な神経発達障害の要因となります。
5. 16番染色体長腕(16q)の異常の深刻な予後
16番染色体の長腕(16q)に生じる異常は、短腕に比べて報告例が少なく、発生した場合は極めて重篤な表現型(症状)を呈することが多いのが特徴です。
遠位部トリソミー(16q24〜)の致命性
長腕の末端領域を含む「遠位部トリソミー」は、多くが親の均衡型転座に起因して発生します。発生学的に極めて深刻な状態を引き起こし、重度の筋緊張低下、著しい発達遅延、そして非常に高い乳児期死亡率と関連しています。
大動脈弓離断や左室低形成といった心臓外科的介入を要する重度奇形のほか、鎖肛(直腸肛門奇形)が特筆して高い割合で発生します。長期間生存するケースは医学文献上でも非常に稀です。
16q24.1欠失:肺胞毛細血管異形成症(ACD/MPV)
16q24.1領域の微小欠失は、FOXF1遺伝子のハプロ不全を引き起こし、「肺静脈のミスアライメントを伴う肺胞毛細血管異形成症(ACD/MPV)」という極めて致命的な先天性肺疾患の直接的な原因となります。
⚠️ 予後に関する重要な事実:ACD/MPVの重症度
出生直後から治療抵抗性の極めて重度な肺高血圧症を発症します。新生児期致死率がほぼ100%に近く、ECMO(体外式膜型人工肺)を使用しても延命は一時的であり、長期的生存の唯一の選択肢は難易度の高い「乳児肺移植」のみとされています。
6. 遺伝カウンセリングとライフステージに応じた集学的支援
ここまで見てきたように、16番染色体異常は「完全トリソミー」か「モザイク」か、「欠失」か「重複」かによって、描かれる生物学的な運命が大きく異なります。
今日、NIPTなどの出生前診断技術の向上により、こうした微小なバリアントを高い解像度で出生前に把握できるようになりました。しかし、それは同時に「不完全浸透」などの曖昧な情報に両親が直面し、深刻な心理的負担を強いられることを意味します。
💡 出生前診断(NIPT)で不安な方へのアプローチ
- ➤リスク因子の正しい解釈:16p重複のように「健常に育つ可能性も高い」ものを、確定的な悲観材料と混同しないこと。
- ➤確定診断へのステップ:NIPTはあくまでスクリーニングです。陽性の場合は、胎児の正確な状況を知るために羊水検査等を検討します。
- ➤集学的医療の準備:モザイクトリソミーなど生存の可能性がある場合、出生直後からのNICU管理や小児神経科、リハビリ等のサポート体制を整えることで、社会への主流化(通常学級等)を強力に後押しできます。
🏥 専門医による遺伝カウンセリングで「次の一手」を
染色体異常の告知は、情報が複雑で不安が増幅しやすいものです。
私たちは最新の遺伝学知見に基づき、正確なリスクの解釈と心の安全を最優先に、次の一手を一緒に整理します。
よくある質問(FAQ)
🧬 その他の染色体異常(トリソミー・部分モノソミー)について
各染色体の異数性や微小欠失・重複による特徴的な疾患、および予後については以下のリンクから詳細をご確認いただけます。
関連記事
参考文献
- [1] Mosaic trisomy 16: what are the obstetric and long-term childhood outcomes? [PMC]
- [2] Trisomy 16 and trisomy 16 Mosaicism: a review [PubMed]
- [3] 16p11.2 Recurrent Deletion – GeneReviews® [NCBI Bookshelf]
- [4] Rubinstein-Taybi Syndrome – GeneReviews® [NCBI Bookshelf]
- [5] Alpha-Thalassemia – GeneReviews® [NCBI Bookshelf]
- [6] Distal duplication 16q syndrome [Orphanet]
- [7] Alveolar Capillary Dysplasia [PMC]






